WO2011028950A1 - Mutant smoothened and methods of using the same - Google Patents
Mutant smoothened and methods of using the same Download PDFInfo
- Publication number
- WO2011028950A1 WO2011028950A1 PCT/US2010/047739 US2010047739W WO2011028950A1 WO 2011028950 A1 WO2011028950 A1 WO 2011028950A1 US 2010047739 W US2010047739 W US 2010047739W WO 2011028950 A1 WO2011028950 A1 WO 2011028950A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- smo
- amino acid
- antibodies
- mutant
- Prior art date
Links
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/74—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving hormones or other non-cytokine intercellular protein regulatory factors such as growth factors, including receptors to hormones and growth factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/72—Assays involving receptors, cell surface antigens or cell surface determinants for hormones
- G01N2333/726—G protein coupled receptor, e.g. TSHR-thyrotropin-receptor, LH/hCG receptor, FSH
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2500/00—Screening for compounds of potential therapeutic value
- G01N2500/10—Screening for compounds of potential therapeutic value involving cells
Definitions
- the present invention relates to isolated mutant SMO nucleic acids and proteins related to chemotherapeutic resistance of tumors and methods of screening for compounds that bind to SMO mutants, or modulate SMO activity, and to cancer diagnostics and therapies and in particular to the detection of mutations that are diagnostic and/or prognostic and treatment of drug-resistant tumors.
- NSCLC EGFR-mutant non-small cell lung cancer
- Medulloblastoma is a primitive neuroectodermal tumor of the cerebellum that represents the most common brain malignancy in children (Polkinghorn, W.R. and N.J. Tarbell (2007) Nat. Clin. Pract. Oncol. 4(5):295-304). Despite improvements in survival rates, the debilitating side effects of adjuvant radiation represent a major clinical challenge, thus supporting the need for new molecular targeted therapies.
- the Hedgehog (Hh) signaling pathway has been directly implicated in the pathogenesis of medulloblastoma. Constitutive Hh signaling, most often due to underlying loss of function mutations in the inhibitory receptor PTCH1, has been demonstrated in approximately 30% of sporadic cases (Zurawel, R.H. et al. (2000) Genes Chromosomes Cancer 27(l):44-51 ; Kool, M. et al. (2008) PLoS ONE
- the invention provides isolated nucleic acid molecules encoding a mutant SMO protein.
- the nucleic acid molecules encode an amino acid sequence that is at least 95% identical to SEQ ID NO:2 wherein said amino acid sequence comprises an amino acid at position 473 of SEQ ID NO:2 that is any amino acid other than aspartic acid (D).
- the amino acid at position 473 of SEQ ID NO:2 is histidine (H), glycine (G), phenylalanine (F), tyrosine (Y), leucine (L), isoleucine (I), proline (P), serine (S), threonine (T), methionine (M), glutamine (Q), or asparagine (N).
- the isolated nucleic acid sequence comprising a parental nucleic acid sequence of SEQ ID NO:3 (wild- type SMO), but containing a mutation or mutations at positions 1417, 1418 and/or
- the mutations result in a change from aspartic acid (D) to histidine (H), glycine (G), phenylalanine (F), tyrosine (Y), leucine (L), isoleucine (I), proline (P), serine (S), threonine (T), methionine (M), glutamine (Q), or asparagine (N).
- the invention provides nucleic acid probes capable of specifically hybridizing to a nucleic acid encoding a mutated SMO protein or fragment thereof incorporating a mutation in amino acid 473 of SMO.
- he probe is complementary to the nucleic acid encoding the mutated SMO or said fragment thereof.
- the probe may have a length of about 10 to about 50 nucleotides.
- the probe may be detectably labeled. The probe differentially binds mutant Smo over wild-type Smo (havind an aspartic acid at position 473).
- the invention also provides an isolated mutant SMO protein comprising an amino acid sequence of that is at least 95% identical to SEQ ID NO:2 wherein the amino acid sequence comprises an amino acid at position 473 other than aspartic acid (D).
- the amino acid at position 473 is histidine (H), glycine (G), phenylalanine (F), tyrosine (Y), leucine (L), isoleucine (I), proline (P), serine (S), threonine (T), methionine (M), glutamine (Q), or asparagine (N).
- the invention further provides an antibody that specifically binds to the mutant SMO protein of the invention wherein the epitope of the antibody is present on a mutant SMO having an amino acid other than aspartic acid at position 473, but does not bind to wild-type SMO.
- the antibody binds with high affinity to mutant SMO, but does not bind with high affinity to wild-type SMO.
- the antibody is a monoclonal antibody, a chimeric antibody, a humanized antibody, a single chain antibody or an antigen-binding fragment thereof (e.g., a Fab, a Fab', a F(ab') 2 , or an Fv fragment).
- the antibody is conjugated to a detectable label.
- the antibody is conjugated to a cytotoxic agent, such as, but not limited to a chemotherapeutic agent, a toxin or a radioactive isotope.
- a cytotoxic agent such as, but not limited to a chemotherapeutic agent, a toxin or a radioactive isotope.
- the antibody inhibits SMO activity. In other embodiments, the antibody inhibits only mutant SMO activity.
- the invention also provides a method of detecting a mutated SMO gene in a sample comprising amplifying from a sample a nucleic acid encoding the carboxy- terminus of transmembrane domain 6 of SMO, or a fragment thereof suspected of containing a mutation, and comparing the electrophoretic mobility of the amplified nucleic acid to the electrophoretic mobility of corresponding wild-type SMO gene or fragment thereof.
- the electrophoretic mobility is determined on polyacrylamide gel.
- the electrophoretic mobility of mutant Smo can be differentiated from wild-type Smo.
- the invention further provides a method of identifying at least one SMO mutation in a sample comprising contacting a nucleic acid from the sample with a nucleic acid probe that is capable of specifically hybridizing to a nucleic acid encoding a mutated SMO protein, or fragment thereof incorporating a mutation, and detecting hybridization.
- the method detects a mutation in the carboxy-terminal portion of transmembrane domain 6 of SMO.
- the SMO mutation occurs in Smo at positions 1417, 1418, and/or 1419 (encoding amino acid at position 473) wherein the mutation results in a codon encoding an amino acid other than aspartic acid.
- the probe is detectably labeled.
- the probe is an antisense oligomer.
- the nucleic acid of the SMO gene or a fragment thereof in the sample is amplified and contacted with the probe.
- the invention also provides a method for identifying a tumor in a human subject that is resistant to treatment with a chemotherapeutic agent such as GDC-0449 comprising determining the presence of a mutated SMO gene or mutated SMO protein in a sample of the tumor wherein said mutation is located in the SMO gene that encodes a portion of SMO at the extracellular membrane surface (e.g. , the carboxy-terminal portion of transmembrane domain 6 of SMO) whereby the presence of the mutated SMO gene or mutated SMO protein indicates that the tumor is resistant to treatment with the chemotherapeutic agent, such as, but not limited to GDC-0449.
- the chemotherapeutic agent is GDC-0449.
- the chemotherapeutic agent is cyclopamine.
- the mutation is in a portion of the SMO gene that encodes amino acid 473 of SMO.
- the mutation causes a change in amino acid 473 of SMO from Asp to another amino acid.
- the other amino acid is histidine (H), glycine (G), phenylalanine (F), tyrosine (Y), leucine (L), isoleucine (I), proline (P), serine (S), threonine (T), methionine (M), glutamine (Q), or asparagine (N).
- the invention also provides a method for identifying a tumor in a human subject that is susceptible to treatment with an SMO inhibitor comprising (i) determining the presence of a wild-type SMO protein or gene in a sample of the tumor whereby the presence of a wild-type SMO protein or gene indicates that the tumor is susceptible to treatment with a SMO inhibitor or (ii) determining the presence of a mutated SMO protein or gene in a sample of the tumor wherein the mutation results in a change of amino acid at position 473 of SMO, whereby the presence of a mutated SMO protein or gene indicates that the tumor is not susceptible to treatment with a SMO inhibitor such as GDC-0449.
- the SMO mutation is a change from aspartic acid (D)473 to any other amino acid.
- the amino acid is histidine (H), glycine (G), phenylalanine (F), tyrosine (Y), leucine (L), isoleucine (I), proline (P), serine (S), threonine (T), methionine (M), glutamine (Q), or asparagine (N).
- the invention also provides a method of determining prognosis of patient being treated for a Hedgehog-dependent tumor comprising determining in a sample of a tumor the presence or absence of a mutation at amino acid 473 whereby the presence of the mutation indicates poorer prognosis compared to the absence of said mutation using certain Smo inhibitors.
- the invention further provides a method of screening for compounds that inhibit signaling of a mutant SMO protein that incorporates a mutation at amino acid 473 comprising contacting the mutant SMO with a test compound and detecting binding of the compound to the mutant SMO whereby binding of the test compound to mutant SMO indicates that the test compound is an inhibitor of mutant SMO.
- the invention also provides a method of screening for compounds that inhibit signaling of a mutant SMO protein that incorporates a mutation at amino acid 473 comprising contacting a cell that expresses the mutant SMO with a test compound and detecting activity of Gli in the cell whereby the presence of Gli activity indicates that the test compound is not an inhibitor of mutant SMO.
- Gli activity is measured using a Gli protein that is conjugated to a detectable label.
- the detectable label is a fluorescent label (e.g., luciferase).
- the invention also provides a method for treating cancer by administering to a patient in need thereof a compound that specifically binds to SMO having an amino acid substitution (mutation) at position 473.
- the mutant SMO protein comprises the substitution from aspartic acid at 473 to any other amino acid.
- the other amino acid is histidine (H), glycine (G),
- the compound is an antibody.
- the compound is a small molecule having the structural formula of Formula I, Formula II and/or Formula III (see below).
- the invention also provides a method for delaying or preventing drug-induced mutagenesis comprising administering an inhibitor of SMO and a PI3K inhibitor.
- the SMO inhibitor is GDC-0449.
- the SMO inhibitor is an inhibitor of a mutant SMO having an amino acid substitution at position 473.
- the mutant SMO inhibitor is a compound having the structural formula of Formula I, Formula II or Formula III (see below).
- Figure 1 shows identification of a SMO mutation in tumor samples from a medulloblastoma patient who relapsed after an initial response to GDC-0449.
- A Nucleotide sequence tracings showing a heterozygous mutation in SMO causing a Asp>His change at amino acid 473 (asterisk). This mutation was present in a metastatic biopsy taken at relapse, but was not present in the primary tumor prior to GDC-0449 treatment.
- B The GPCR architecture of SMO maps the location of the D473H mutation to the C-terminal end of TM6.
- FIG. 2 shows The SMO D473H mutation confers resistance to the Hh pathway inhibitor GDC-0449.
- A GLI-luciferase reporter activity after transfection of SMO variants in the presence (grey bars) or absence (black bars) of PTCH1 DNA (20ng).
- SMO-M2 represents a previously identified activating mutation.
- B GLI- luciferase reporter activity in C3H10T1/2 cells transfected with SMO-WT (closed circles) or SMO-D473H (open circles) after treatment with various doses of GDC- 0449. Reporter activity is normalized to untreated cultures.
- Figure 3 shows acquired resistance to GDC-0449 through SMO mutation in a genetically-engineered mouse model of medulloblastoma.
- A Medulloblastoma allografts from Ptch+/-;p53-/- mice were made GDC-0449 resistant through intermittent daily dosing with 75 mg/kg GDC-0449. Treatment days are represented by triangles and tumors were excised once they failed to respond to twice daily dosing with GDC-0449.
- B Nucleotide sequence tracings from parental and a GDC-0449- resistant (SG274) medulloblastoma allografts showing a heterozygous mutation resulting in a D>G change at amino acid 477 of SMO (homologous to pos.
- C GLI-luciferase reporter activity in C3H10T1/2 cells transfected with SMO-WT (closed circles) or SMO-D477G (open circles) after treatment with various doses of GDC-0449.
- Figure 4 shows the presence and loss of heterozygosity (LOH) of the preexisting PTCH1 W844C mutation is confirmed in the biopsy taken at relapse.
- Nucleotide sequence tracings confirm the pre-existing PTCH1 W844C homozygous mutation in a biopsy taken at relapse.
- B Loss of heterozygosity on chromosome 9 in DNA obtained from the biopsy at relapse, as assessed by AffymetrixSNP arrays. Stretches of homozygous allele calls for each SNP probe across the highlighted region of chromosome 9 are shown.
- Figure 5 shows PTCH1-W844C is unable to suppress Hh pathway activity.
- GLI-luciferase reporter activity following co-transfection of various input ratios of SMO and either WT (closed circles) or W844C (open circles) PTCH1 DNA in C3H10T1/2 cells.
- Figure 6 shows no SMO copy number alterations were detected by qPCR using 2 independent assays from gDNA derived from the biopsy at progression. Copy number was determined by qPCR and calibrated to normal human genomic DNA following normalization to LINE-1. As controls, gDNA from cell lines with low-level copy number changes at the SMO locus, as determined previously by
- Frzreceptors An alignment across the TM6-TM7 region of representative SMO species variants and the ten human Frz receptor chains shows the conserved Asp/Glu residue at position 473. The TM7 tail position of Trp-535 that harbors the SMO-M2 activating mutation is also highlighted. Interestingly, both sensitive amino acid positions are closely followed by a short, membrane-associated amphipathic helix.
- B assessment of niGlil mRNA levels by qRT-PCR in tumors from panel (A) collected 6 hours after the last drug treatment. Values represent means ⁇ SDs.
- C representative images of S12 cells treated with indicated compounds in the absence (top) or presence (bottom) of Shh for 16 hours. Cilia and centrosomes (acetylated and gamma tubulins respectively, as well as Smo were detected by immunofluorescence, while nuclei were visualized by DAPI staining.
- Figure 17 shows that control and GDC-0449-resistant MB allografts are sensitive to PI3K inhibition.
- B mean fitted tumor volumes of control and GDC-0449-resistant MB allografts treated orally with either vehicle (open squares) or 150 mg kg-1 GDC- 0941 once daily (solid triangles).
- an “isolated” antibody is one which has been identified and separated and/or recovered from a component of its natural environment. Contaminant components of its natural environment are materials which would interfere with research, diagnostic or therapeutic uses for the antibody, and may include enzymes, hormones, and other proteinaceous or nonproteinaceous solutes.
- an antibody is purified (1) to greater than 95% by weight of antibody as determined by, for example, the Lowry method, and in some embodiments, to greater than 99% by weight; (2) to a degree sufficient to obtain at least 15 residues of N-terminal or internal amino acid sequence by use of, for example, a spinning cup sequenator, or (3) to homogeneity by SDS-PAGE under reducing or nonreducing conditions using, for example, Coomassie blue or silver stain.
- Isolated antibody includes the antibody in situ within
- “Native antibodies” are usually heterotetrameric glycoproteins of about 150,000 daltons, composed of two identical light (L) chains and two identical heavy (H) chains. Each light chain is linked to a heavy chain by one covalent disulfide bond, while the number of disulfide linkages varies among the heavy chains of different immunoglobulin isotypes. Each heavy and light chain also has regularly spaced intrachain disulfide bridges. Each heavy chain has at one end a variable domain (V R ) followed by a number of constant domains.
- V R variable domain
- Each light chain has a variable domain at one end (V L ) and a constant domain at its other end; the constant domain of the light chain is aligned with the first constant domain of the heavy chain, and the light chain variable domain is aligned with the variable domain of the heavy chain. Particular amino acid residues are believed to form an interface between the light chain and heavy chain variable domains.
- variable region or “variable domain” of an antibody refers to the amino- terminal domains of the heavy or light chain of the antibody.
- variable domain of the heavy chain may be referred to as "VH.”
- variable domain of the light chain may be referred to as "VL.” These domains are generally the most variable parts of an antibody and contain the antigen-binding sites.
- variable refers to the fact that certain portions of the variable domains differ extensively in sequence among antibodies and are used in the binding and specificity of each particular antibody for its particular antigen. However, the variability is not evenly distributed throughout the variable domains of antibodies. It is concentrated in three segments called hypervariable regions (HVRs) both in the light-chain and the heavy-chain variable domains. The more highly conserved portions of variable domains are called the framework regions (FR).
- HVRs hypervariable regions
- FR framework regions
- the variable domains of native heavy and light chains each comprise four FR regions, largely adopting a beta-sheet configuration, connected by three HVRs, which form loops connecting, and in some cases forming part of, the beta-sheet structure.
- the HVRs in each chain are held together in close proximity by the FR regions and, with the HVRs from the other chain, contribute to the formation of the antigen-binding site of antibodies (see Kabat et al., Sequences of Proteins of Immunological Interest, Fifth Edition, National Institute of Health, Bethesda, MD (1991)).
- the constant domains are not involved directly in the binding of an antibody to an antigen, but exhibit various effector functions, such as participation of the antibody in antibody-dependent cellular toxicity.
- the "light chains" of antibodies (immunoglobulins) from any vertebrate species can be assigned to one of two clearly distinct types, called kappa ( ⁇ ) and lambda ( ⁇ ), based on the amino acid sequences of their constant domains.
- antibodies immunoglobulins
- antibodies can be assigned to different classes.
- full length antibody “intact antibody” and “whole antibody” are used herein interchangeably to refer to an antibody in its substantially intact form, not antibody fragments as defined below.
- Antibody fragments comprise a portion of an intact antibody, preferably comprising the antigen binding region thereof.
- antibody fragments include Fab, Fab', F(ab') 2 , and Fv fragments; diabodies; linear antibodies; single-chain antibody molecules; and multispecific antibodies formed from antibody fragments.
- Papain digestion of antibodies produces two identical antigen-binding fragments, called “Fab” fragments, each with a single antigen-binding site, and a residual "Fc” fragment, whose name reflects its ability to crystallize readily. Pepsin treatment yields an F(ab') 2 fragment that has two antigen-combining sites and is still capable of cross-linking antigen.
- the Fab fragment contains the heavy- and light-chain variable domains and also contains the constant domain of the light chain and the first constant domain (CHI) of the heavy chain.
- Fab' fragments differ from Fab fragments by the addition of a few residues at the carboxy terminus of the heavy chain CHI domain including one or more cysteines from the antibody hinge region.
- Fab'-SH is the designation herein for Fab' in which the cysteine residue(s) of the constant domains bear a free thiol group.
- F(ab') 2 antibody fragments originally were produced as pairs of Fab' fragments which have hinge cysteines between them. Other chemical couplings of antibody fragments are also known.
- Single-chain Fv or “scFv” antibody fragments comprise the VH and VL domains of antibody, wherein these domains are present in a single polypeptide chain.
- the scFv polypeptide further comprises a polypeptide linker between the VH and VL domains which enables the scFv to form the desired structure for antigen binding.
- diabodies refers to antibody fragments with two antigen-binding sites, which fragments comprise a heavy-chain variable domain (VH) connected to a light-chain variable domain (VL) in the same polypeptide chain (VH-VL).
- VH heavy-chain variable domain
- VL light-chain variable domain
- Diabodies may be bivalent or bispecific. Diabodies are described more fully in, for example, EP 404,097; WO 1993/01161; Hudson et al., Nat. Med. 9: 129-134 (2003); and Hollinger et al, Proc. Natl. Acad. Sci. USA 90: 6444-6448 (1993). Triabodies and tetrabodies are also described in Hudson et al, Nat. Med. 9: 129-134 (2003).
- the selection process can be the selection of a unique clone from a plurality of clones, such as a pool of hybridoma clones, phage clones, or recombinant DNA clones.
- a selected target binding sequence can be further altered, for example, to improve affinity for the target, to humanize the target binding sequence, to improve its production in cell culture, to reduce its immunogenicity in vivo, to create a multispecific antibody, etc., and that an antibody comprising the altered target binding sequence is also a monoclonal antibody of this invention.
- polyclonal antibody In contrast to polyclonal antibody
- each monoclonal antibody of a monoclonal antibody preparation is directed against a single determinant on an antigen.
- monoclonal antibody preparations are advantageous in that they are typically uncontaminated by other immunoglobulins.
- the modifier "monoclonal” indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method.
- the monoclonal antibodies to be used in accordance with the present invention may be made by a variety of techniques, including, for example, the hybridoma method ⁇ e.g., Kohler and Milstein, Nature, 256:495-97 (1975); Hongo et al, Hybridoma, 14 (3): 253-260 (1995), Harlow et al., Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory Press, 2nd ed.
- the monoclonal antibodies herein specifically include "chimeric" antibodies in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in antibodies derived from a particular species or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in antibodies derived from another species or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity (see, e.g.,U.S. Patent No. 4,816,567; and Morrison et al, Proc. Natl. Acad. Sci. USA 81 :6851-6855 (1984)).
- Chimeric antibodies include PRIMATIZED® antibodies wherein the antigen-binding region of the antibody is derived from an antibody produced by, e.g., immunizing macaque monkeys with the antigen of interest.
- Humanized forms of non-human (e.g., murine) antibodies are chimeric antibodies that contain minimal sequence derived from non-human immunoglobulin.
- a humanized antibody is a human immunoglobulin (recipient antibody) in which residues from a HVR of the recipient are replaced by residues from a HVR of a non-human species (donor antibody) such as mouse, rat, rabbit, or nonhuman primate having the desired specificity, affinity, and/or capacity.
- donor antibody such as mouse, rat, rabbit, or nonhuman primate having the desired specificity, affinity, and/or capacity.
- FR residues of the human immunoglobulin are replaced by corresponding non-human residues.
- humanized antibodies may comprise residues that are not found in the recipient antibody or in the donor antibody. These modifications may be made to further refine antibody performance.
- a humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin, and all or substantially all of the FRs are those of a human immunoglobulin sequence.
- the humanized antibody optionally will also comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin.
- Fc immunoglobulin constant region
- a "human antibody” is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human and/or has been made using any of the techniques for making human antibodies as disclosed herein. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues. Human antibodies can be produced using various techniques known in the art, including phage-display libraries.
- Human antibodies can be prepared by administering the antigen to a transgenic animal that has been modified to produce such antibodies in response to antigenic challenge, but whose endogenous loci have been disabled, e.g., immunized xenomiee (see, e.g., U.S. Pat. Nos. 6,075,181 and 6,150,584 regarding XENOMOUSETM technology). See also, for example, Li et al, Proc. Natl. Acad.
- HVR hypervariable region
- VL VL
- H3 and L3 display the most diversity of the six HVRs, and H3 in particular is believed to play a unique role in conferring fine specificity to antibodies.
- HVR delineations are in use and are encompassed herein.
- the Kabat Complementarity Determining Regions are based on sequence variability and are the most commonly used (Kabat et al, Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD. (1991)). Chothia refers instead to the location of the structural loops (Chothia and Lesk J. Mol. Biol. 196:901-917 (1987)).
- the AbM HVRs represent a compromise between the Kabat HVRs and Chothia structural loops, and are used by Oxford Molecular's AbM antibody modeling software.
- the "contact" HVRs are based on an analysis of the available complex crystal structures. The residues from each of these HVRs are noted below.
- Framework or "FR” residues are those variable domain residues other than the HVR residues as herein defined.
- the Kabat numbering system is generally used when referring to a residue in the variable domain (approximately residues 1-107 of the light chain and residues 1- 113 of the heavy chain) (e.g, Kabat et al., Sequences of Immunological Interest. 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)).
- the "EU numbering system” or "EU index” is generally used when referring to a residue in an immunoglobulin heavy chain constant region ⁇ e.g., the EU index reported in Kabat et al., supra).
- the "EU index as in Kabat” refers to the residue numbering of the human IgGl EU antibody.
- references to residue numbers in the variable domain of antibodies means residue numbering by the Kabat numbering system. Unless stated otherwise herein, references to residue numbers in the constant domain of antibodies means residue numbering by the EU numbering system ⁇ e.g., see United States Provisional Application No. 60/640,323, Figures for EU numbering).
- blocking antibody or an “antagonist” antibody is one which inhibits or reduces biological activity of the antigen it binds. Certain blocking antibodies or antagonist antibodies substantially or completely inhibit the biological activity of the antigen.
- “Growth inhibitory” antibodies are those that prevent or reduce proliferation of a cell expressing an antigen to which the antibody binds.
- the antibody may prevent or reduce proliferation of cancer cells that express Smo or mutant in vitro and/or in vivo.
- a “native sequence Fc region” comprises an amino acid sequence identical to the amino acid sequence of an Fc region found in nature.
- Native sequence human Fc regions include a native sequence human IgGl Fc region (non-A and A allotypes); native sequence human IgG2 Fc region; native sequence human IgG3 Fc region; and native sequence human IgG4 Fc region as well as naturally occurring variants thereof.
- a “variant Fc region” comprises an amino acid sequence which differs from that of a native sequence Fc region by virtue of at least one amino acid modification, preferably one or more amino acid substitution(s).
- the variant Fc region has at least one amino acid substitution compared to a native sequence Fc region or to the Fc region of a parent polypeptide, e.g. from about one to about ten amino acid substitutions, and preferably from about one to about five amino acid substitutions in a native sequence Fc region or in the Fc region of the parent polypeptide.
- Fc receptor or “FcR” describes a receptor that binds to the Fc region of an antibody.
- an FcR is a native human FcR.
- Inhibiting receptor FcyRIIB contains an immunoreceptor tyrosine-based inhibition motif (ITIM) in its cytoplasmic domain, (see, e.g., Daeron, Annu. Rev. Immunol. 15:203-234 (1997)).
- ITIM immunoreceptor tyrosine-based inhibition motif
- FcRs are reviewed, for example, in Ravetch and Kinet, Annu. Rev. Immunol 9:457-92 (1991); Capel et al, Immunomethods 4:25-34 (1994); and de Haas et al, J. Lab. Clin. Med. 126:330-41 (1995).
- Other FcRs including those to be identified in the future, are encompassed by the term "FcR" herein.
- Fc receptor or “FcR” also includes the neonatal receptor, FcRn, which is responsible for the transfer of maternal IgGs to the fetus (Guyer et al, J. Immunol. 117:587 (1976) and Kim et al, J. Immunol. 24:249 (1994)) and regulation of homeostasis of immunoglobulins. Methods of measuring binding to FcRn are known (see, e.g., Ghetie and Ward., Immunol. Today 18(12):592-598 (1997); Ghetie et al, Nature Biotechnology, 15(7):637-640 (1997); Hinton et al., J. Biol. Chem. 279(8):6213-6216 (2004); WO 2004/92219 (Hinton et al).
- Human effector cells are leukocytes which express one or more FcRs and perform effector functions. In certain embodiments, the cells express at least FcyRIII and perform ADCC effector function(s). Examples of human leukocytes which mediate ADCC include peripheral blood mononuclear cells (PBMC), natural killer (NK) cells, monocytes, cytotoxic T cells, and neutrophils.
- PBMC peripheral blood mononuclear cells
- NK natural killer cells
- monocytes cytotoxic T cells
- neutrophils neutrophils.
- the effector cells may be isolated from a native source, e.g. , from blood.
- ADCC activity of a molecule of interest may be assessed in vitro, such as that described in US Patent No. 5,500,362 or 5,821,337 or U.S. Patent No. 6,737,056 (Presta).
- Useful effector cells for such assays include PBMC and NK cells.
- ADCC activity of the molecule of interest may be assessed in vivo, e.g., in an animal model such as that disclosed in Clynes et al. PNAS (USA) 95:652-656 (1998).
- Fc region-comprising antibody refers to an antibody that comprises an Fc region.
- the C-terminal lysine (residue 447 according to the EU numbering system) of the Fc region may be removed, for example, during purification of the antibody or by recombinant engineering of the nucleic acid encoding the antibody.
- a composition comprising an antibody having an Fc region according to this invention can comprise an antibody with K447, with all K447 removed, or a mixture of antibodies with and without the K447 residue.
- the "Kd" or "Kd value” according to this invention is measured by a radiolabeled antigen binding assay (RIA) performed with the Fab version of an antibody of interest and its antigen as described by the following assay.
- RIA radiolabeled antigen binding assay
- Solution binding affinity of Fabs for antigen is measured by equilibrating Fab with a minimal concentration of ( 125 I)-labeled antigen in the presence of a titration series of unlabeled antigen, then capturing bound antigen with an anti-Fab antibody-coated plate (see, e.g., Chen, et al., J. Mol. Biol. 293:865-881(1999)).
- MICROTITER ® multi-well plates (Thermo Scientific) are coated overnight with 5 ⁇ g/ml of a capturing anti-Fab antibody (Cappel Labs) in 50 mM sodium carbonate (pH 9.6), and subsequently blocked with 2% (w/v) bovine serum albumin in PBS for two to five hours at room temperature (approximately
- the Kd or Kd value is measured by using surface plasmon resonance assays using a BIACORE ® -2000 or a BIACORE ® -3000 (BIAcore, Inc., Piscataway, NJ) at 25°C with immobilized antigen CM5 chips at -10 response units (RU).
- CM5 carboxymethylated dextran biosensor chips
- EDC N-ethyl-N'- (3-dimethylaminopropyl)- carbodiimide hydrochloride
- NHS N-hydroxysuccinimide
- Antigen is diluted with 10 mM sodium acetate, pH 4.8, to 5 ⁇ g/ml (-0.2 ⁇ ) before injection at a flow rate of 5 ⁇ /minute to achieve approximately 10 response units (RU) of coupled protein. Following the injection of antigen, 1 M ethanolamine is injected to block unreacted groups. For kinetics measurements, two-fold serial dilutions of Fab (0.78 nM to 500 nM) are injected in PBS with 0.05% TWEEN-20TM surfactant (PBST) at 25°C at a flow rate of approximately 25 ⁇ /min. Association rates (k on ) and dissociation rates (k 0 ff) are calculated using a simple one-to-one Langmuir binding model (BIACORE ®
- a spectrometer such as a stop-flow equipped spectrophometer (Aviv Instruments) or a 8000-series SLM-AMINCOTM spectrophotometer (ThermoSpectronic) with a stirred cuvette.
- a spectrometer such as a stop-flow equipped spectrophometer (Aviv Instruments) or a 8000-series SLM-AMINCOTM spectrophotometer (ThermoSpectronic) with a stirred cuvette.
- an “on-rate,” “rate of association,” “association rate,” or “k on” can also be determined as described above using a BIACORE ® -2000 or a BIACORE ® -3000 system (BIAcore, Inc., Piscataway, NJ).
- substantially similar denotes a sufficiently high degree of similarity between two numeric values (for example, one associated with an antibody of the invention and the other associated with a reference/comparator antibody), such that one of skill in the art would consider the difference between the two values to be of little or no biological and/or statistical significance within the context of the biological characteristic measured by said values (e.g., Kd values).
- the difference between said two values is, for example, less than about 50%, less than about 40%, less than about 30%>, less than about 20%>, and/or less than about 10% as a function of the reference/comparator value.
- the difference between the two values is, for example, greater than about 10%>, greater than about 20%, greater than about 30%, greater than about 40%, and/or greater than about 50% as a function of the value for the reference/comparator molecule.
- “Purified” means that a molecule is present in a sample at a concentration of at least 95%) by weight, or at least 98%> by weight of the sample in which it is contained.
- nucleic acid molecule is a nucleic acid molecule that is separated from at least one other nucleic acid molecule with which it is ordinarily associated, for example, in its natural environment.
- An isolated nucleic acid molecule further includes a nucleic acid molecule contained in cells that ordinarily express the nucleic acid molecule, but the nucleic acid molecule is present extrachromosomally or at a chromosomal location that is different from its natural chromosomal location.
- vector is intended to refer to a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked.
- plasmid refers to a circular double stranded DNA into which additional DNA segments may be ligated.
- phage vector refers to a viral vector, wherein additional DNA segments may be ligated into the viral genome.
- viral vector capable of autonomous replication in a host cell into which they are introduced (e.g., bacterial vectors having a bacterial origin of replication and episomal mammalian vectors).
- vectors e.g., non-episomal mammalian vectors
- vectors can be integrated into the genome of a host cell upon introduction into the host cell, and thereby are replicated along with the host genome.
- certain vectors are capable of directing the expression of genes to which they are operatively linked.
- Such vectors are referred to herein as "recombinant expression vectors," or simply, "expression vectors.”
- expression vectors of utility in recombinant DNA techniques are often in the form of plasmids.
- plasmid and vector may be used interchangeably as the plasmid is the most commonly used form of vector.
- Polynucleotide or “nucleic acid,” as used interchangeably herein, refer to polymers of nucleotides of any length, and include DNA and R A.
- the nucleotides can be deoxyribonucleotides, ribonucleotides, modified nucleotides or bases, and/or their analogs, or any substrate that can be incorporated into a polymer by DNA or RNA polymerase or by a synthetic reaction.
- a polynucleotide may comprise modified nucleotides, such as methylated nucleotides and their analogs. If present, modification to the nucleotide structure may be imparted before or after assembly of the polymer.
- the sequence of nucleotides may be interrupted by non-nucleotide components.
- a polynucleotide may comprise modification(s) made after synthesis, such as conjugation to a label.
- modifications include, for example, "caps," substitution of one or more of the naturally occurring nucleotides with an analog, internucleotide modifications such as, for example, those with uncharged linkages (e.g., methyl phosphonates, phosphotriesters, phosphoamidates, carbamates, etc.) and with charged linkages (e.g., phosphorothioates, phosphorodithioates, etc.), those containing pendant moieties, such as, for example, proteins (e.g., nucleases, toxins, antibodies, signal peptides, ply-L-lysine, etc.), those with intercalators (e.g., acridine, psoralen, etc.), those containing chelators (e.g., metals,
- any of the hydroxyl groups ordinarily present in the sugars may be replaced, for example, by phosphonate groups, phosphate groups, protected by standard protecting groups, or activated to prepare additional linkages to additional nucleotides, or may be conjugated to solid or semi-solid supports.
- the 5' and 3' terminal OH can be phosphorylated or substituted with amines or organic capping group moieties of from 1 to 20 carbon atoms.
- Other hydroxyls may also be derivatized to standard protecting groups.
- Polynucleotides can also contain analogous forms of ribose or deoxyribose sugars that are generally known in the art, including, for example, 2'-0-methyl-, 2'-0-allyl-, 2'-fluoro- or 2'-azido-ribose, carbocyclic sugar analogs, a-anomeric sugars, epimeric sugars such as arabinose, xyloses or lyxoses, pyranose sugars, furanose sugars, sedoheptuloses, acyclic analogs, and basic nucleoside analogs such as methyl riboside.
- One or more phosphodiester linkages may be replaced by alternative linking groups.
- linking groups include, but are not limited to, embodiments wherein phosphate is replaced by P(0)S ("thioate”), P(S)S ("dithioate”), (0)NR 2 ("amidate”), P(0)R, P(0)OR', CO, or CH2 ("formacetal”), in which each R or R' is independently H or substituted or
- unsubstituted alkyl (1-20 C) optionally containing an ether (-0-) linkage, aryl, alkenyl, cycloalkyl, cycloalkenyl or araldyl. Not all linkages in a polynucleotide need be identical. The preceding description applies to all polynucleotides referred to herein, including RNA and DNA.
- Oligonucleotide generally refers to short, generally single- stranded, generally synthetic polynucleotides that are generally, but not necessarily, less than about 200 nucleotides in length.
- oligonucleotide and
- polynucleotide are not mutually exclusive. The description above for
- polynucleotides is equally and fully applicable to oligonucleotides.
- SMO refers to any native SMO from any vertebrate source, including mammals such as primates (e.g. humans) and rodents (e.g., mice and rats), unless otherwise indicated.
- the term encompasses "full-length,” unprocessed SMO as well as any form of SMO that results from processing in the cell.
- the term also encompasses naturally occurring variants of SMO, e.g., splice variants or allelic variants.
- “Mutant Smo” as used herein refers to SMO having a mutation in the carboxy-terminal portion of transmembrane 6 of SMO at position 473 of human SMO.
- treatment refers to clinical intervention in an attempt to alter the natural course of the individual or cell being treated, and can be performed either for prophylaxis or during the course of clinical pathology. Desirable effects of treatment include preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, preventing metastasis, decreasing the rate of disease progression, amelioration or palliation of the disease state, and remission or improved prognosis.
- antibodies of the invention are used to delay development of a disease or disorder or to slow the progression of a disease or disorder.
- An “individual,” “subject,” or “patient” is a vertebrate.
- the vertebrate is a mammal.
- Mammals include, but are not limited to, farm animals (such as cows), sport animals, pets (such as cats, dogs, and horses), primates, mice and rats.
- a mammal is a human.
- pharmaceutical formulation refers to a preparation which is in such form as to permit the biological activity of the active ingredient to be effective, and which contains no additional components which are unacceptably toxic to a subject to which the formulation would be administered. Such formulations may be sterile.
- a "sterile" formulation is aseptic or free from all living microorganisms and their spores.
- an “effective amount” refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired therapeutic or prophylactic result.
- a “therapeutically effective amount” of a substance/molecule of the invention may vary according to factors such as the disease state, age, sex, and weight of the individual, and the ability of the substance/molecule, to elicit a desired response in the individual.
- a therapeutically effective amount encompasses an amount in which any toxic or detrimental effects of the substance/molecule are outweighed by the therapeutically beneficial effects.
- a “prophylactically effective amount” refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired prophylactic result. Typically, but not necessarily, since a prophylactic dose is used in subjects prior to or at an earlier stage of disease, the prophylactically effective amount would be less than the therapeutically effective amount.
- cytotoxic agent refers to a substance that inhibits or prevents a cellular function and/or causes cell death or destruction.
- the term is intended to include radioactive isotopes (e.g., At 211 , 1 131 , 1 125 , Y 90 , Re 186 , Re 188 , Sm 153 ,
- chemotherapeutic agents e.g., methotrexate, adriamicin, vinca alkaloids (vincristine, vinblastine, etoposide), doxorubicin, melphalan, mitomycin C, chlorambucil, daunorubicin or other intercalating agents, enzymes and fragments thereof such as nucleolytic enzymes, antibiotics, and toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including fragments and/or variants thereof, and the various antitumor or anticancer agents disclosed below.
- Other cytotoxic agents are described below.
- a tumoricidal agent causes destruction of tumor cells.
- a "toxin” is any substance capable of having a detrimental effect on the growth or proliferation of a cell.
- chemotherapeutic agent is a chemical compound useful in the treatment of cancer.
- examples of chemotherapeutic agents include alkylating agents such as thiotepa and cyclosphosphamide (CYTOXAN®); alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine,
- triethylenemelamine triethylenephosphoramide, triethylenethiophosphoramide and trimethylomelamine
- acetogenins especially bullatacin and bullatacinone
- delta-9- tetrahydrocannabinol (dronabinol, MARINOL®); beta-lapachone
- lapachol lapachol
- colchicines include betulinic acid; a camptothecin (including the synthetic analogue topotecan (HYCAMTIN®), CPT-11 (irinotecan, CAMPTOSAR®),
- camptothecin including the synthetic analogue topotecan (HYCAMTIN®), CPT-11 (irinotecan, CAMPTOSAR®)
- chlorophosphamide estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosoureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, and ranimnustine; antibiotics such as the enediyne antibiotics (e. g., calicheamicin, especially calicheamicin gammall and calicheamicin omegall (see, e.g., Nicolaou et al., Angew. Chem Intl. Ed.
- folic acid analogues such as denopterin, methotrexate, pteropterin, trimetrexate;
- purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti- adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid;
- eniluracil amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elfornithine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidainine; maytansinoids such as maytansine and
- ansamitocins mitoguazone; mitoxantrone; mopidanmol; nitraerine; pentostatin;
- TAXOL® paclitaxel
- ABRAXANETM albumin-engineered nanoparticle formulation of paclitaxel
- TXOTERE® docetaxel
- chloranbucil 6-thioguanine; mercaptopurine; methotrexate; platinum agents such as cisplatin, oxaliplatin (e.g., ELOXATIN®), and carboplatin; vincas, which prevent tubulin polymerization from forming microtubules, including vinblastine
- platinum agents such as cisplatin, oxaliplatin (e.g., ELOXATIN®), and carboplatin
- vincas which prevent tubulin polymerization from forming microtubules, including vinblastine
- VELBAN® vincristine
- ELDISINE® vindesine
- ELDISINE® FILDESIN®
- NAVELBINE® etoposide
- ifosfamide mitoxantrone; leucovorin; novantrone; edatrexate; daunomycin; aminopterin; ibandronate;
- topoisomerase inhibitor RFS 2000 difluoromethylornithine (DMFO); retinoids such as retinoic acid, including bexarotene (TARGRETIN®); bisphosphonates such as clodronate (for example, BONEFOS® or OSTAC®), etidronate (DIDROCAL®), NE-58095, zoledronic acid/zoledronate (ZOMETA®), alendronate (FOSAMAX®), pamidronate (AREDIA®), tiludronate (SKELID®), or risedronate (ACTONEL®); troxacitabine (a 1,3-dioxolane nucleoside cytosine analog); antisense
- clodronate for example, BONEFOS® or OSTAC®
- etidronate etidronate
- ZOMETA® alendronate
- AREDIA® pamidronate
- SKELID® tiludronate
- oligonucleotides particularly those that inhibit expression of genes in signaling pathways implicated in aberrant cell proliferation, such as, for example, PKC-alpha, Raf, H-Ras, and epidermal growth factor receptor (EGF-R); vaccines such as THERATOPE® vaccine and gene therapy vaccines, for example, ALLOVECTIN® vaccine, LEUVECTIN® vaccine, and VAXID® vaccine; topoisomerase 1 inhibitor (e.g., LURTOTECAN®); rmRH (e.g., ABARELIX®); BAY439006 (sorafenib;
- SU- 1 1248 (sunitinib, SUTENT®, Pfizer); perifosine, COX-2 inhibitor (e.g. celecoxib or etoricoxib), proteosome inhibitor (e.g. PS341); bortezomib
- VELCADE® CCI-779; tipifarnib (R1 1577); orafenib, ABT510; Bcl-2 inhibitor such as oblimersen sodium (GENASENSE®); pixantrone; EGFR inhibitors (see definition below); tyrosine kinase inhibitors (see definition below); serine-threonine kinase inhibitors such as rapamycin (sirolimus, RAPAMUNE®); farnesyltransferase inhibitors such as lonafarnib (SCH 6636, SARASARTM); and pharmaceutically acceptable salts, acids or derivatives of any of the above; as well as combinations of two or more of the above such as CHOP, an abbreviation for a combined therapy of cyclophosphamide, doxorubicin, vincristine, and prednisolone; and FOLFOX, an abbreviation for a treatment regimen with oxaliplatin (ELOXATINTM) combined
- Chemotherapeutic agents as defined herein include “anti-hormonal agents” or “endocrine therapeutics” which act to regulate, reduce, block, or inhibit the effects of hormones that can promote the growth of cancer. They may be hormones themselves, including, but not limited to: anti-estrogens with mixed agonist/antagonist profile, including, tamoxifen (NOLVADEX®), 4-hydroxytamoxifen, toremifene
- SERM3 selective estrogen receptor modulators
- pure anti-estrogens without agonist properties such as fulvestrant (FASLODEX®), and EM800 (such agents may block estrogen receptor (ER) dimerization, inhibit DNA binding, increase ER turnover, and/or suppress ER levels); aromatase inhibitors, including steroidal aromatase inhibitors such as formestane and exemestane
- aromatase inhibitors include vorozole (RIVISOR®), megestrol acetate (MEGASE®), fadrozole, and 4(5)-imidazoles; lutenizing hormone-releaseing hormone agonists, including leuprolide (LUPRON® and ELIGARD®), goserelin, buserelin, and tripterelin; sex steroids, including progestines such as megestrol acetate and medroxyprogesterone acetate, estrogens such as diethylstilbestrol and premarin, and androgens/retinoids such as fluoxymesterone, all transretionic acid and fenretinide; onapristone; anti- progesterones; estrogen receptor down-regulators (ERDs); anti-androgens such as flutamide, nilutamide and bicalutamide; and pharmaceutically acceptable salt
- a “growth inhibitory agent” when used herein refers to a compound or composition which inhibits growth of a cell (such as a cell expressing SMO) either in vitro or in vivo.
- the growth inhibitory agent may be one which significantly reduces the percentage of cells (such as a cell expressing SMO) in S phase.
- growth inhibitory agents include agents that block cell cycle progression (at a place other than S phase), such as agents that induce Gl arrest and M-phase arrest.
- Classical M-phase blockers include the vincas (vincristine and vinblastine), taxanes, and topoisomerase II inhibitors such as doxorubicin, epirubicin, daunorubicin, etoposide, and bleomycin.
- Those agents that arrest Gl also spill over into S-phase arrest, for example, DNA alkylating agents such as tamoxifen, prednisone, dacarbazine, mechlorethamine, cisplatin, methotrexate, 5-fluorouracil, and ara-C.
- DNA alkylating agents such as tamoxifen, prednisone, dacarbazine, mechlorethamine, cisplatin, methotrexate, 5-fluorouracil, and ara-C.
- antineoplastic drugs by Murakami et al. (W.B. Saunders, Philadelphia, 1995), e.g., p. 13.
- the taxanes are anticancer drugs both derived from the yew tree.
- Docetaxel (TAXOTERE®, Rhone-Poulenc Rorer), derived from the
- European yew is a semisynthetic analogue of paclitaxel (TAXOL®, Bristol-Myers Squibb). Paclitaxel and docetaxel promote the assembly of microtubules from tubulin dimers and stabilize microtubules by preventing depolymerization, which results in the inhibition of mitosis in cells.
- a “mutant Smo antagonist” is a compound that inhibits the biological activity of a SMO having an amino acid substitution at position 473 of human SMO that changes the wild-type aspartic acid at this position to any other amino acid.
- the biological activity of SMO is the ability to transducer a signal upon stimulation with hedgehog to activation of Gli transcription factor.
- the nucleic acids of the invention include isolated mutant SMO-encoding sequences.
- the nucleic acids comprise a sequence that is at least 80% identical to the nucleic acid sequence of SEQ ID NO: 3 and which contain at least one mutation from this sequence to encode any amino acid at position 473 other than aspartic acid (D).
- the nucleic acid encodes a histidine (H), glycine (G), phenylalanine (F), tyrosine (Y), leucine (L), isoleucine (I), proline (P), serine (S), threonine (T), methionine (M), glutamine (Q), or asparagine (N) at position 473.
- the nucleic acid has at least one mutation from the parental wild- type SMO at nucleotide 1417, 1418 and/or 1419.
- the percent identity is 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% with SEQ ID NO:3 providing that there is at least one mutation at position 1417, 1418 and/or 1419.
- the invention also contemplates fragments of such nucleic acids that span the region of the mutations described above in fragments that are at least 20 nucleotides in length.
- the nucleotide fragments are 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, or 100 nucleotides in length.
- the fragments may be any length that spans the region of the mutations described above up to the full length mutant SMO-encoding nucleic acid molecule.
- Isolated mutant SMO and fragments thereof may be used, for example, for hybridization, to generate primers and probes for the prognostic and diagnostic assays of the invention, and for expression in recombinant systems (such as to generate mutant SMO protein or portions thereof for use as immunogens and for use in assays of the invention as described herein).
- the invention provides nucleic acid probes which may be used to identify the mutant SMO nucleic acid molecule in the methods of the invention.
- Nucleic acid samples derived from tissue suspected of having a mutant SMO or from tissue wherein the status of SMO is unknown may be screened using a specific probe for mutant SMO using standard procedures, such as described in Sambrook et al., MOLECULAR CLONING: A LABORATORY MANUAL, Cold Spring Harbor Laboratory Press, NY, 1989).
- the nucleic acid encoding SMO may be amplified from the tissue and probed with a specific probe of the invention to determine the presence of absence of mutant SMO.
- PCR methodology is well known in the art (Sambrook et al, supra; Dieffenbach et al, PCR PRIMER: A LABORATORY MANUAL, Cold Spring Harbor Laboratory Press, NY, 1995).
- Nucleotide sequences (or their complement) encoding mutant SMO have various applications in the art of molecular biology, including uses as hybridization probes, and in the generation of anti-sense RNA and DNA probes. Mutant SMO- encoding nucleic acid will also be useful for the preparation of mutant SMO polypeptides by the recombinant techniques described herein, wherein those mutant SMO polypeptides may find use, for example, in the preparation of anti-mutant SMO antibodies as described herein.
- the full-length mutant SMO nucleic acids, or portions thereof, may be used as hybridization probes for identifying mutant SMO.
- the length of the probes will be about 20 to about 50 bases.
- the hybridization probes may be derived from at least the mutant region of the full length mutant SNO nucleotide sequence.
- a screening method will comprise isolating the coding region of mutant SMO using the known DNA sequence to synthesize a selected probe of about 40 bases.
- Hybridization probes may be labeled by a variety of labels,
- Labeled probes having a sequence complementary to that of the mutant SMO gene of the present invention can be used to screen libraries of human cDNA, genomic DNA or mRNA to determine which members of such libraries the probe hybridizes to.
- Hybridization products may be resolved on polyacrylamide gels.
- the SMO mutations may be determined using the method described in the Examples. Hybridization conditions, including moderate stringency and high stringency, are provided in Sambrook et al., supra.
- Sequences identified in such library screening methods can be compared and aligned to the known sequences for SMO and mutant SMO. Sequence identity at the carboxy-terminal region of transmembrane domain 6 can be determined using methods known in the art.
- antisense or sense oligonucleotides comprising a single-stranded nucleic acid sequence (either RNA or DNA) capable of binding to target mutant SMO mRNA (sense) or mutant SMO DNA (antisense) sequences.
- Antisense or sense oligonucleotides comprise a fragment of the coding region of mutant SMO DNA containing the mutation region. Such a fragment generally comprises at least about 14 nucleotides, preferably from about 14 to 30 nucleotides.
- binding of antisense or sense oligonucleotides to target nucleic acid sequences results in the formation of duplexes that block transcription or translation of the target sequence by one of several means, including enhanced degradation of the duplexes, premature termination of transcription or translation, or by other means.
- the antisense oligonucleotides thus may be used to block expression of mutant SMO proteins, wherein those mutant SMO proteins may play a role in the resistance of cancer in mammals to
- Antisense or sense oligonucleotides further comprise oligonucleotides having modified sugar-phosphodiester backbones (or other sugar linkages, such as those described in WO 91/06629) and wherein such sugar linkages are resistant to endogenous nucleases.
- Such oligonucleotides with resistant sugar linkages are stable in vivo (i.e., capable of resisting enzymatic degradation) but retain sequence specificity to be able to bind to target nucleotide sequences.
- oligonucleotides containing modified backbones or non-natural internucleoside linkages include those that retain a phosphorus atom in the backbone and those that do not have a phosphorus atom in the backbone.
- modified oligonucleotides that do not have a phosphorus atom in their internucleoside backbone can also be considered to be oligonucleosides.
- Preferred modified oligonucleotide backbones include, for example, phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkylphosphotri-esters, methyl and other alkyl phosphonates including 3'-alkylene phosphonates, 5'-alkylene phosphonates and chiral
- Preferred oligonucleotides having inverted polarity comprise a single 3' to 3' linkage at the 3'- most internucleotide linkage i.e. a single inverted nucleoside residue which may be abasic (the nucleobase is missing or has a hydroxyl group in place thereof).
- Various salts, mixed salts and free acid forms are also included.
- Representative United States patents that teach the preparation of phosphorus-containing linkages include, but are not limited to, U.S. Patent Nos.: 3,687,808; 4,469,863; 4,476,301; 5,023,243;
- Preferred modified oligonucleotide backbones that do not include a phosphorus atom therein have backbones that are formed by short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl internucleoside linkages, or one or more short chain heteroatomic or heterocyclic internucleoside linkages.
- nucleobases are retained and are bound directly or indirectly to aza nitrogen atoms of the amide portion of the backbone.
- Representative United States patents that teach the preparation of PNA compounds include, but are not limited to, U.S. Patent Nos.: 5,539,082; 5,714,331; and 5,719,262, each of which is herein incorporated by reference. Further teaching of PNA compounds can be found in Nielsen et a/.(1991) Science 254: 1497-1500.
- a preferred modification includes 2'-methoxyethoxy (2'-0-CH 2 CH 2 0CH 3 , also known as 2'-0-(2- methoxyethyl) or 2'-MOE) (Martin et al. (1995) Helv. Chim. Acta 78:486-504) i.e., an alkoxyalkoxy group.
- a further preferred modification includes 2'- dimethylaminooxyethoxy, i.e., a 0(CH 2 ) 2 0N(CH 3 ) 2 group, also known as 2'-DMAOE, as described in examples hereinbelow, and 2'-dimethylaminoethoxyethoxy (also known in the art as 2'-0-dimethylaminoethoxyethyl or 2'-DMAEOE), i.e., 2'-0-CH 2 - 0-CH 2 -N(CH 2 ).
- a further preferred modification includes Locked Nucleic Acids (LNAs) in which the 2'-hydroxyl group is linked to the 3' or 4' carbon atom of the sugar ring thereby forming a bicyclic sugar moiety.
- the linkage is preferably a methelyne (- CH 2 -) n group bridging the 2' oxygen atom and the 4' carbon atom wherein n is 1 or 2.
- LNAs and preparation thereof are described in WO 98/39352 and WO 99/14226.
- the 2'-modification may be in the arabino (up) position or ribo (down) position.
- a preferred 2'-arabino modification is 2'-F.
- Similar modifications may also be made at other positions on the oligonucleotide, particularly the 3' position of the sugar on the 3' terminal nucleotide or in 2'-5' linked
- oligonucleotides and the 5' position of 5' terminal nucleotide. Oligonucleotides may also have sugar mimetics such as cyclobutyl moieties in place of the pentofuranosyl sugar.
- Representative U.S. patents that teach the preparation of such modified sugar structures include, but are not limited to, U.S. Patent Nos.: 4,981,957; 5,118,800; 5,319,080; 5,359,044; 5,393,878; 5,446,137; 5,466,786; 5,514,785; 5,519,134;
- Oligonucleotides may also include nucleobase (often referred to in the art simply as “base”) modifications or substitutions.
- nucleobases include the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
- nucleobases include tricyclic pyrimidines such as phenoxazine cytidine (1H- pyrimido[5,4-b][l,4]benzoxazin-2(3H)-one), phenothiazine cytidine (1H- pyrimido[5,4-b][l,4]benzothiazin-2(3H)-one), G-clamps such as a substituted phenoxazine cytidine (e.g., 9-(2-aminoethoxy)-H-pyrimido[5,4-b][l,4]benzoxazin- 2(3H)-one), carbazole cytidine (2H-pyrimido[4,5-b]indol-2-one), pyridoindole cytidine (H-pyrido[3',2':4,5]pyrrolo[2,3-d]pyrimidin-2-one).
- Modified nucleobases may also include those in which the purine or pyrimidine base is replaced with other heterocycles, for example 7-deaza-adenine, 7-deazaguanosine, 2-aminopyridine and 2-pyridone.
- Further nucleobases include those disclosed in U.S. Patent No. 3,687,808, those disclosed in THE CONCISE ENCYCLOPEDIA OF POLYMER SCIENCE AND
- nucleobases are particularly useful for increasing the binding affinity of the oligomeric compounds of the invention. These include 5 -substituted pyrimidines, 6-azapyrimidines and N-2, N-6 and 0-6 substituted purines, including 2-aminopropyl adenine, 5-propynyluracil and 5- propynylcytosine. 5-methylcytosine substitutions have been shown to increase nucleic acid duplex stability by 0.6-1.2 °C. (Sanghvi et al. ANTISENSE RESEARCH AND
- the compounds of the invention can include conjugate groups covalently bound to functional groups such as primary or secondary hydroxyl groups.
- Conjugate groups of the invention include intercalators, reporter molecules, polyamines, polyamides, polyethylene glycols, polyethers, groups that enhance the pharmacodynamic properties of oligomers, and groups that enhance the pharmacokinetic properties of oligomers.
- Typical conjugates groups include cholesterols, lipids, cation lipids, phospholipids, cationic
- pharmacodynamic properties include groups that improve oligomer uptake, enhance oligomer resistance to degradation, and/or strengthen sequence-specific hybridization with R A.
- Groups that enhance the pharmacokinetic properties include groups that improve oligomer uptake, distribution, metabolism or excretion.
- Conjugate moieties include but are not limited to lipid moieties such as a cholesterol moiety (Letsinger et al. (1989) Proc. Natl. Acad. Sci. USA 86:6553-6556), cholic acid (Manoharan et al. (1994) Bioorg. Med. Chem. Lett.
- Oligonucleotides of the invention may also be conjugated to active drug substances, for example, aspirin, warfarin, phenylbutazone, ibuprofen, suprofen, fenbufen, ketoprofen, (S)-(+)-pranoprofen, carprofen, dansylsarcosine, 2,3,5-triiodobenzoic acid, flufenamic acid, folinic acid, a benzothiadiazide, chlorothiazide, a diazepine, indomethicin, a barbiturate, a cephalosporin, a sulfa drug, an antidiabetic, an antibacterial or an antibiotic. Oligonucleotide-drug conjugates and their preparation are described in U.S. Patent Nos.: 4,828,979; 4,948,882; 5,218,105; 5,525,465;
- Chimeric antisense compounds of the invention may be formed as composite structures of two or more oligonucleotides, modified oligonucleotides,
- Such compounds have also been referred to in the art as hybrids or gapmers.
- Preferred gapmers have a region of 2' modified sugars (preferably 2'-0-(CH 2 ) 2 -0-CH 3 ) at the 3'- terminal and at the 5' terminal separated by at least one region having at least 4 contiguous 2'-H sugars and preferably incorporate phosphorothioate backbone linkages.
- Representative United States patents that teach the preparation of such hybrid structures include, but are not limited to, U.S. Patent Nos.: 5,013,830;
- the compounds of the invention may also be admixed, encapsulated, conjugated or otherwise associated with other molecules, molecule structures or mixtures of compounds, as for example, liposomes, receptor targeted molecules, oral, rectal, topical or other formulations, for assisting in uptake, distribution and/or absorption.
- Representative United States patents that teach the preparation of such uptake, distribution and/or absorption assisting formulations include, but are not limited to, U.S. Patent Nos.: 5,108,921; 5,354,844; 5,416,016; 5,459,127; 5,521,291; 5,543,158; 5,547,932; 5,583,020; 5,591,721; 4,426,330;
- oligonucleotide for a target nucleic acid sequence such as poly-(L-lysine).
- intercalating agents such as ellipticine, and alkylating agents or metal complexes may be attached to sense or antisense oligonucleotides to modify binding specificities of the antisense or sense oligonucleotide for the target nucleotide sequence.
- Antisense or sense oligonucleotides may be introduced into a cell containing the target nucleic acid sequence by any gene transfer method, including, for example, CaP0 4 -mediated DNA transfection, electroporation, or by using gene transfer vectors such as Epstein-Barr virus.
- an antisense or sense oligonucleotide is inserted into a suitable retroviral vector.
- a cell containing the target nucleic acid sequence is contacted with the recombinant retroviral vector, either in vivo or ex vivo.
- Suitable retroviral vectors include, but are not limited to, those derived from the murine retrovirus M-MuLV, N2 (a retrovirus derived from M- MuLV), or the double copy vectors designated DCT5A, DCT5B and DCT5C (see WO 90/13641).
- a sense or an antisense oligonucleotide may be introduced into a cell containing the target nucleic acid sequence by formation of an oligonucleotide- lipid complex, as described in WO 90/10448.
- the sense or antisense oligonucleotide- lipid complex is preferably dissociated within the cell by an endogenous lipase.
- Antisense or sense R A or DNA molecules are generally at least about 5 nucleotides in length, alternatively at least about 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, 500, 510, 520, 530, 540, 550, 560, 570, 580, 590, 600, 610, 620, 630,
- Nucleotide sequences encoding a mutant SMO can also be used to construct hybridization probes for mapping the gene which encodes that SMO and for the genetic analysis of individuals with genetic disorders.
- the nucleotide sequences provided herein may be mapped to a chromosome and specific regions of a chromosome using known techniques, such as in situ hybridization, linkage analysis against known chromosomal markers, and hybridization screening with libraries.
- a potential mutant SMO antagonist is an antisense RNA or DNA construct prepared using antisense technology, where, e.g. , an antisense RNA or DNA molecule acts to block directly the translation of mRNA by hybridizing to targeted mRNA and preventing protein translation.
- Antisense technology can be used to control gene expression through triple-helix formation or antisense DNA or RNA, both of which methods are based on binding of a polynucleotide to DNA or RNA.
- nucleic acids encoding mutant SMO herein are used to design an antisense RNA oligonucleotide of from about 10 to 40 base pairs in length.
- oligonucleotides described above can also be delivered to cells such that the antisense RNA or DNA may be expressed in vivo to inhibit production of the mutant SMO.
- antisense DNA oligodeoxyribonucleotides derived from the translation-initiation site, e.g., between about -10 and +10 positions of the target gene nucleotide sequence, are preferred.
- the base composition of these oligonucleotides is designed such that it promotes triple-helix formation via Hoogsteen base-pairing rules, which generally require sizeable stretches of purines or pyrimidines on one strand of a duplex.
- Hoogsteen base-pairing rules which generally require sizeable stretches of purines or pyrimidines on one strand of a duplex.
- small molecules can be identified by any one or more of the screening assays discussed hereinabove and/or by any other screening techniques well known for those skilled in the art.
- Examples of the small molecules that may be used as mutant SMO antagonists are compounds having the following structural formulas:
- the invention provides isolated mutant SMO proteins. Wild-type human SMO is shown in SEQ ID NO: 1.
- Mutant human SMO is shown in SEQ ID NO:2 wherein amino acid 473 is shown as "X" which, with respect to this application stands for any amino acid other than aspartic acid (D).
- the X is histidine (H), glycine (G), phenylalanine (F), tyrosine (Y), leucine (L), isoleucine (I), proline (P), serine (S), threonine (T), methionine (M), glutamine (Q), or asparagine (N).
- Mutant SMO and fragments thereof may be produced in recombinant systems as is well known in the art using the mutant SMO nucleic acids described herein. Such nucleic acids may be incorporated into expression vectors as are well-known in that art and transfected into host cells, which may be prokaryotic or eukaryotic cells depending on the proposed use of the protein. Full length or fragments of mutant SMO (in which the fragments contain at least the carboxy-terminal portion of transmembrane domain 6 and amino acid 473 of SEQ ID NO:2) may be used as immunogens to produce antibodies of the invention, or to purify antibodies of the invention, for example.
- an anti-SMO antibody is a monoclonal antibody.
- an anti-SMO antibody is an antibody fragment, e.g., a Fab, Fab'- SH, Fv, scFv, or (Fab') 2 fragment.
- an anti-mutant SMO antibody is a chimeric, humanized, or human antibody.
- an anti-SMO antibody is purified.
- a composition is a pharmaceutical formulation for the treatment of cancer.
- the present invention encompasses antibody fragments.
- Antibody fragments may be generated by traditional means, such as enzymatic digestion, or by
- Fab'-SH fragments can be directly recovered from E. coli and chemically coupled to form F(ab') 2 fragments (Carter et al, Bio/Technology 10: 163- 167 (1992)).
- F(ab') 2 fragments can be isolated directly from recombinant host cell culture.
- Fab and F(ab') 2 fragment with increased in vivo half-life comprising salvage receptor binding epitope residues are described in U.S. Pat. No. 5,869,046.
- Other techniques for the production of antibody fragments will be apparent to the skilled practitioner.
- an antibody is a single chain Fv fragment (scFv). See WO 93/16185; U.S. Pat. Nos. 5,571,894; and
- the invention encompasses humanized antibodies.
- Various methods for humanizing non-human antibodies are known in the art.
- a humanized antibody can have one or more amino acid residues introduced into it from a source which is non-human. These non-human amino acid residues are often referred to as "import" residues, which are typically taken from an "import" variable domain.
- Humanization can be essentially performed following the method of Winter and coworkers (Jones et al. (1986) Nature 321 :522-525; Riechmann et al. (1988) Nature 332:323-327; Verhoeyen et al. (1988) Science 239: 1534-1536), by substituting hypervariable region sequences for the corresponding sequences of a human antibody.
- such "humanized" antibodies are chimeric antibodies (U.S. Patent No. 4,816,567) wherein substantially less than an intact human variable domain has been substituted by the corresponding sequence from a non-human species.
- humanized antibodies are typically human antibodies in which some hypervariable region residues and possibly some FR residues are substituted by residues from analogous sites in rodent antibodies.
- variable domains both light and heavy
- the choice of human variable domains, both light and heavy, to be used in making the humanized antibodies can be important to reduce antigenicity.
- sequence of the variable domain of a rodent antibody is screened against the entire library of known human variable-domain sequences.
- the human sequence which is closest to that of the rodent is then accepted as the human framework for the humanized antibody. See, e.g., Sims et al. (1993) J. Immunol. 151 :2296; Chothia et al. (1987) J. Mol. Biol. 196:901.
- Another method uses a particular framework derived from the consensus sequence of all human antibodies of a particular subgroup of light or heavy chains.
- the same framework may be used for several different humanized antibodies. See, e.g., Carter et al. (1992) Proc. Natl. Acad. Sci. USA, 89:4285; Presta et al. (1993) J. Immunol, 151 :2623.
- humanized antibodies are prepared by a process of analysis of the parental sequences and various conceptual humanized products using three-dimensional models of the parental and humanized sequences.
- Three- dimensional immunoglobulin models are commonly available and are familiar to those skilled in the art.
- Computer programs are available which illustrate and display probable three-dimensional conformational structures of selected candidate immunoglobulin sequences. Inspection of these displays permits analysis of the likely role of the residues in the functioning of the candidate immunoglobulin sequence, i.e., the analysis of residues that influence the ability of the candidate immunoglobulin to bind its antigen.
- FR residues can be selected and combined from the recipient and import sequences so that the desired antibody characteristic, such as increased affinity for the target antigen(s), is achieved.
- the hypervariable region residues are directly and most substantially involved in influencing antigen binding.
- Gene shuffling can also be used to derive human antibodies from non-human, e.g. rodent, antibodies, where the human antibody has similar affinities and specificities to the starting non-human antibody.
- this method which is also called “epitope imprinting"
- either the heavy or light chain variable region of a non-human antibody fragment obtained by phage display techniques as described herein is replaced with a repertoire of human V domain genes, creating a population of non-human chain/human chain scFv or Fab chimeras.
- bispecific antibodies are based on the co-expression of two immunoglobulin heavy chain-light chain pairs, where the two heavy chains have different specificities (Milstein and Cuello, Nature, 305: 537 (1983)). Because of the random assortment of immunoglobulin heavy and light chains, these hybridomas
- the bispecific antibodies are composed of a hybrid immunoglobulin heavy chain with a first binding specificity in one arm, and a hybrid immunoglobulin heavy chain-light chain pair (providing a second binding specificity) in the other arm. It was found that this asymmetric structure facilitates the separation of the desired bispecific compound from unwanted immunoglobulin chain combinations, as the presence of an immunoglobulin light chain in only one half of the bispecific molecule provides for a facile way of separation. This approach is disclosed in WO 94/04690. For further details of generating bispecific antibodies see, for example, Suresh et al, Methods in Enzymology, 121 :210 (1986).
- the interface between a pair of antibody molecules can be engineered to maximize the percentage of heterodimers which are recovered from recombinant cell culture.
- the interface comprises at least a part of the C H 3 domain of an antibody constant domain.
- one or more small amino acid side chains from the interface of the first antibody molecule are replaced with larger side chains (e.g. tyrosine or tryptophan).
- Compensatory "cavities" of identical or similar size to the large side chain(s) are created on the interface of the second antibody molecule by replacing large amino acid side chains with smaller ones (e.g. alanine or threonine). This provides a mechanism for increasing the yield of the heterodimer over other unwanted end-products such as homodimers.
- Antibodies with more than two valencies are contemplated.
- trispecific antibodies can be prepared. Tutt et al J. Immunol 147: 60 (1991).
- a multivalent antibody may be internalized (and/or catabolized) faster than a bivalent antibody by a cell expressing an antigen to which the antibodies bind.
- the antibodies of the present invention can be multivalent antibodies (which are other than of the IgM class) with three or more antigen binding sites (e.g. tetravalent antibodies), which can be readily produced by recombinant expression of nucleic acid encoding the polypeptide chains of the antibody.
- the multivalent antibody can comprise a dimerization domain and three or more antigen binding sites.
- the dimerization domain comprises (or consists of) an Fc region or a hinge region. In this scenario, the antibody will comprise an Fc region and three or more antigen binding sites amino-terminal to the Fc region.
- a multivalent antibody comprises (or consists of) three to about eight antigen binding sites. In one such embodiment, a multivalent antibody comprises (or consists of) four antigen binding sites.
- the multivalent antibody comprises at least one polypeptide chain (for example, two polypeptide chains), wherein the polypeptide chain(s) comprise two or more variable domains.
- the polypeptide chain(s) may comprise VD1- (Xl)n -VD2-(X2)n -Fc, wherein VD1 is a first variable domain, VD2 is a second variable domain, Fc is one polypeptide chain of an Fc region, XI and X2 represent an amino acid or polypeptide, and n is 0 or 1.
- an antibody of the invention is a single-domain antibody.
- a single-domain antibody is a single polyeptide chain comprising all or a portion of the heavy chain variable domain or all or a portion of the light chain variable domain of an antibody.
- a single-domain antibody is a human single-domain antibody (Domantis, Inc., Waltham, MA; see, e.g., U.S. Patent No. 6,248,516 Bl).
- a single-domain antibody consists of all or a portion of the heavy chain variable domain of an antibody.
- amino acid sequence modification(s) of the antibodies described herein are contemplated.
- Amino acid sequence variants of the antibody may be prepared by introducing appropriate changes into the nucleotide sequence encoding the antibody, or by peptide synthesis. Such modifications include, for example, deletions from, and/or insertions into and/or substitutions of, residues within the amino acid sequences of the antibody. Any combination of deletion, insertion, and substitution can be made to arrive at the final construct, provided that the final construct possesses the desired characteristics.
- the amino acid alterations may be introduced in the subject antibody amino acid sequence at the time that sequence is made.
- a useful method for identification of certain residues or regions of the antibody that are preferred locations for mutagenesis is called "alanine scanning mutagenesis" as described by Cunningham and Wells (1989) Science, 244: 1081-1085.
- a residue or group of target residues are identified ⁇ e.g., charged residues such as arg, asp, his, lys, and glu) and replaced by a neutral or negatively charged amino acid ⁇ e.g., alanine or polyalanine) to affect the interaction of the amino acids with antigen.
- Those amino acid locations demonstrating functional sensitivity to the substitutions then are refined by introducing further or other variants at, or for, the sites of substitution.
- the site for introducing an amino acid sequence variation is predetermined, the nature of the mutation per se need not be
- ala scanning or random mutagenesis is conducted at the target codon or region and the expressed immunoglobulins are screened for the desired activity.
- Amino acid sequence insertions include amino- and/or carboxyl-terminal fusions ranging in length from one residue to polypeptides containing a hundred or more residues, as well as intrasequence insertions of single or multiple amino acid residues.
- terminal insertions include an antibody with an N-terminal methionyl residue.
- Other insertional variants of the antibody molecule include the fusion to the N- or C-terminus of the antibody to an enzyme ⁇ e.g. for ADEPT) or a polypeptide which increases the serum half-life of the antibody.
- an antibody of the invention is altered to increase or decrease the extent to which the antibody is glycosylated.
- Glycosylation of polypeptides is typically either N-linked or O-linked.
- N-linked refers to the attachment of a carbohydrate moiety to the side chain of an asparagine residue.
- the tripeptide sequences asparagine-X-serine and asparagine-X-threonine, where X is any amino acid except proline, are the recognition sequences for enzymatic attachment of the carbohydrate moiety to the asparagine side chain.
- the presence of either of these tripeptide sequences in a polypeptide creates a potential glycosylation site.
- O- linked glycosylation refers to the attachment of one of the sugars N- aceylgalactosamine, galactose, or xylose to a hydroxyamino acid, most commonly serine or threonine, although 5-hydroxyproline or 5-hydroxylysine may also be used.
- Addition or deletion of glycosylation sites to the antibody is conveniently accomplished by altering the amino acid sequence such that one or more of the above- described tripeptide sequences (for N-linked glycosylation sites) is created or removed. The alteration may also be made by the addition, deletion, or substitution of one or more serine or threonine residues to the sequence of the original antibody (for O-linked glycosylation sites).
- the carbohydrate attached thereto may be altered.
- Native antibodies produced by mammalian cells typically comprise a branched, biantennary oligosaccharide that is generally attached by an N-linkage to Asn297 of the CH2 domain of the Fc region. See, e.g., Wright et al. (1997) TIBTECH 15:26-32.
- the oligosaccharide may include various carbohydrates, e.g., mannose, N- acetyl glucosamine (GlcNAc), galactose, and sialic acid, as well as a fucose attached to a GlcNAc in the "stem" of the biantennary oligosaccharide structure.
- modifications of the oligosaccharide in an antibody of the invention may be made in order to create antibody variants with certain improved properties.
- antibody variants are provided having a carbohydrate structure that lacks fucose attached (directly or indirectly) to an Fc region. Such variants may have improved ADCC function. See, e.g., US Patent Publication Nos. US
- WO2002/031140 Okazaki et al. J. Mol. Biol. 336: 1239-1249 (2004); Yamane- Ohnuki et al. Biotech. Bioeng. 87: 614 (2004).
- Examples of cell lines capable of producing defucosylated antibodies include Led 3 CHO cells deficient in protein fucosylation (Ripka et al. Arch. Biochem. Biophys.
- knockout cell lines such as alpha- 1,6- fucosyltransferase gene, FUT8, knockout CHO cells (see, e.g., Yamane-Ohnuki et al. Biotech. Bioeng. 87: 614 (2004); Kanda, Y. et al, Biotechnol. Bioeng., 94(4):680-688 (2006); and WO2003/085107).
- Antibodies variants are further provided with bisected oligosaccharides, e.g., in which a biantennary oligosaccharide attached to the Fc region of the antibody is bisected by GlcNAc. Such antibody variants may have reduced fucosylation and/or improved ADCC function. Examples of such antibody variants are described, e.g., in WO 2003/011878 (Jean-Mairet et al.); US Patent No. 6,602,684 (Umana et al.); and US 2005/0123546 (Umana et al.). Antibody variants with at least one galactose residue in the oligosaccharide attached to the Fc region are also provided.
- Such antibody variants may have improved CDC function.
- Such antibody variants are described, e.g., in WO 1997/30087 (Patel et al.); WO 1998/58964 (Raju, S.); and WO 1999/22764 (Raju, S.).
- an antibody variant comprises an Fc region with one or more amino acid substitutions which further improve ADCC, for example, substitutions at positions 298, 333, and/or 334 of the Fc region (Eu numbering of residues). Such substitutions may occur in combination with any of the variations described above.
- the invention contemplates an antibody variant that possesses some but not all effector functions, which make it a desirable candidate for many applications in which the half life of the antibody in vivo is important yet certain effector functions (such as complement and ADCC) are unnecessary or deleterious.
- the Fc activities of the antibody are measured to ensure that only the desired properties are maintained.
- In vitro and/or in vivo cytotoxicity assays can be conducted to confirm the reduction/depletion of CDC and/or ADCC activities.
- Fc receptor (FcR) binding assays can be conducted to ensure that the antibody lacks FcyR binding (hence likely lacking ADCC activity), but retains FcRn binding ability.
- NK cells express FcyRIII only, whereas monocytes express FcyRI, FcyRII and FcyRIII.
- FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol. 9:457-92 (1991).
- Non-limiting examples of in vitro assays to assess ADCC activity of a molecule of interest is described in U.S. Patent No. 5,500,362 (see, e.g. Hellstrom, I., et al. Proc. Nat 7 Acad. Sci. USA 83:7059-7063 (1986)) and Hellstrom, I et al., Proc.
- non-radioactive assays methods may be employed (see, for example, ACTITM non-radioactive cytotoxicity assay for flow cytometry
- PBMC peripheral blood mononuclear cells
- NK Natural Killer
- ADCC activity of the molecule of interest may be assessed in vivo, e.g., in a animal model such as that disclosed in Clynes et al. Proc. Nat 'lAcad. Sci. USA 95:652-656 (1998).
- Clq binding assays may also be carried out to confirm that the antibody is unable to bind Clq and hence lacks CDC activity.
- a CDC assay may be performed (see, for example, Gazzano-Santoro et al., J. Immunol. Methods 202: 163 (1996); Cragg, M.S. et al., Blood 101 : 1045-1052 (2003); and Cragg, M.S. and M.J. Glennie, Blood 103:2738- 2743 (2004)).
- FcRn binding and in vivo clearance/half life determinations can also be performed using methods known in the art (see, for example, Petkova, S.B. et al., Int 'l. Immunol. 18(12): 1759-1769 (2006)).
- Sites of interest for substitutional mutagenesis include the hypervariable regions, but FR alterations are also contemplated.
- Conservative substitutions are shown in Table 1 under the heading of "preferred substitutions.” More substantial changes, denominated "exemplary substitutions" are provided in Table 1, or as further described below in reference to amino acid classes.
- Amino acid substitutions may be introduced into an antibody of interest and the products screened, e.g. , for a desired activity, such as improved antigen binding, decreased immunogenicity, improved ADCC or CDC, etc.
- Modifications in the biological properties of an antibody may be accomplished by selecting substitutions that affect (a) the structure of the polypeptide backbone in the area of the substitution, for example, as a sheet or helical conformation, (b) the charge or hydrophobicity of the molecule at the target site, or (c) the bulk of the side chain.
- Amino acids may be grouped according to similarities in the properties of their side chains (in A. L. Lehninger, in Biochemistry, second ed., pp. 73-75, Worth Publishers, New York (1975)):
- Non-conservative substitutions will entail exchanging a member of one of these classes for another class. Such substituted residues also may be introduced into the conservative substitution sites or, into the remaining (non-conserved) sites.
- substitutional variant involves substituting one or more hypervariable region residues of a parent antibody (e.g. a humanized or human antibody).
- a parent antibody e.g. a humanized or human antibody
- the resulting variant(s) selected for further development will have modified (e.g., improved) biological properties relative to the parent antibody from which they are generated.
- An exemplary substitutional variant is an affinity matured antibody, which may be conveniently generated using phage display-based affinity maturation techniques. Briefly, several hypervariable region sites (e.g. 6-7 sites) are mutated to generate all possible amino acid substitutions at each site.
- the antibodies thus generated are displayed from filamentous phage particles as fusions to at least part of a phage coat protein (e.g., the gene III product of Ml 3) packaged within each particle.
- the phage-displayed variants are then screened for their biological activity (e.g. binding affinity).
- scanning mutagenesis e.g., alanine scanning
- contact residues and neighboring residues are candidates for substitution according to techniques known in the art, including those elaborated herein.
- Nucleic acid molecules encoding amino acid sequence variants of the antibody are prepared by a variety of methods known in the art. These methods include, but are not limited to, isolation from a natural source (in the case of naturally occurring amino acid sequence variants) or preparation by oligonucleotide -mediated (or site- directed) mutagenesis, PCR mutagenesis, and cassette mutagenesis of an earlier prepared variant or a non-variant version of the antibody.
- the Fc region variant may comprise a human Fc region sequence (e.g., a human IgGl, IgG2, IgG3 or IgG4 Fc region) comprising an amino acid modification (e.g. a substitution) at one or more amino acid positions including that of a hinge cysteine.
- a human Fc region sequence e.g., a human IgGl, IgG2, IgG3 or IgG4 Fc region
- an amino acid modification e.g. a substitution
- an antibody of the invention may comprise one or more alterations as compared to the wild type counterpart antibody, e.g. in the Fc region. These antibodies would nonetheless retain substantially the same characteristics required for therapeutic utility as compared to their wild type counterpart. For example, it is thought that certain alterations can be made in the Fc region that would result in altered (i.e., either improved or diminished) Clq binding and/or Complement Dependent Cytotoxicity (CDC), e.g., as described in
- modifications facilitate and/or promote heterodimerization. These modifications comprise introduction of a protuberance into a first Fc polypeptide and a cavity into a second Fc polypeptide, wherein the protuberance is positionable in the cavity so as to promote complexing of the first and second Fc polypeptides.
- Methods of generating antibodies with these modifications are known in the art, e.g., as described in U.S. Pat. No. 5,731,168.
- cysteine engineered antibodies e.g., "thioMAbs”
- one or more residues of an antibody are substituted with cysteine residues.
- the substituted residues occur at accessible sites of the antibody.
- reactive thiol groups are thereby positioned at accessible sites of the antibody and may be used to conjugate the antibody to other moieties, such as drug moieties or linker-drug moieties, as described further herein.
- any one or more of the following residues may be substituted with cysteine: V205 (Kabat numbering) of the light chain; Al 18 (EU numbering) of the heavy chain; and S400 (EU numbering) of the heavy chain Fc region.
- the antibodies of the present invention can be further modified to contain additional nonproteinaceous moieties that are known in the art and readily available.
- the moieties suitable for derivatization of the antibody are water soluble polymers.
- water soluble polymers include, but are not limited to, polyethylene glycol (PEG), copolymers of ethylene glycol/propylene glycol, carboxymethylcellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone, poly-1, 3-dioxolane, poly-l,3,6-trioxane, ethylene/maleic anhydride copolymer, polyaminoacids (either homopolymers or random copolymers), and dextran or poly(n- vinyl pyrrolidone)polyethylene glycol, propropylene glycol homopolymers, prolypropylene oxide/ethylene oxide co-polymers, polyoxyethylated polyols (e.g., glycerol), polyvinylene glycol,
- propionaldehyde may have advantages in manufacturing due to its stability in water.
- the polymer may be of any molecular weight, and may be branched or unbranched.
- the number of polymers attached to the antibody may vary, and if more than one polymer are attached, they can be the same or different molecules. In general, the number and/or type of polymers used for derivatization can be determined based on considerations including, but not limited to, the particular properties or functions of the antibody to be improved, whether the antibody derivative will be used in a therapy under defined conditions, etc.
- conjugates of an antibody and nonproteinaceous moiety that may be selectively heated by exposure to radiation are provided.
- the nonproteinaceous moiety is a carbon nanotube (Kam et al, Proc. Natl. Acad. Sci. USA 102: 11600-11605 (2005)).
- the radiation may be of any wavelength, and includes, but is not limited to, wavelengths that do not harm ordinary cells, but which heat the nonproteinaceous moiety to a temperature at which cells proximal to the antibody-nonproteinaceous moiety are killed.
- Monoclonal antibodies of the invention can be made using the hybridoma method first described by Kohler et al, Nature, 256:495 (1975), and further described, e.g., in Hongo et al, Hybridoma, 14 (3): 253-260 (1995), Harlow et al, Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory Press, 2nd ed. 1988); Hammerling et al, in: Monoclonal Antibodies and T-Cell Hybridomas 563- 681 (Elsevier, N.Y., 1981), and Ni, Xiandai Mianyixue, 26(4):265-268 (2006) regarding human-human hybridomas.
- Additional methods include those described, for example, in U.S. Pat. No. 7,189,826 regarding production of monoclonal human natural IgM antibodies from hybridoma cell lines.
- Human hybridoma technology Trioma technology
- Vollmers and Brandlein Histology and Histopathology, 20(3):927-937 (2005)
- Vollmers and Brandlein Methods and Findings in Experimental and Clinical Pharmacology, 27(3): 185-91 (2005).
- Antibodies are raised in animals by multiple subcutaneous (sc) or intraperitoneal (ip) injections of a polypeptide comprising mutant SMO or a fragment thereof, and an adjuvant, such as monophosphoryl lipid A (MPL)/trehalose
- sc subcutaneous
- ip intraperitoneal
- MPL monophosphoryl lipid A
- TDM dicrynomycolate
- Ribi Immunochem. Research, Inc. Hamilton, MT
- a polypeptide comprising mutant SMO or a fragment thereof may be prepared using methods well known in the art, such as recombinant methods, some of which are further described herein. Serum from immunized animals is assayed for anti-mutant SMO antibodies, and booster immunizations are optionally administered.
- Lymphocytes from animals producing anti-mutant SMO antibodies are isolated.
- lymphocytes may be immunized in vitro.
- Lymphocytes are then fused with myeloma cells using a suitable fusing agent, such as polyethylene glycol, to form a hybridoma cell.
- a suitable fusing agent such as polyethylene glycol
- Myeloma cells may be used that fuse efficiently, support stable high-level production of antibody by the selected antibody-producing cells, and are sensitive to a medium such as HAT medium.
- Exemplary myeloma cells include, but are not limited to, murine myeloma lines, such as those derived from MOPC-21 and MPC-11 mouse tumors available from the Salk Institute Cell Distribution Center, San Diego, California USA, and SP-2 or X63-Ag8-653 cells available from the American Type Culture Collection, Rockville, Maryland USA.
- Human myeloma and mouse-human heteromyeloma cell lines also have been described for the production of human monoclonal antibodies (Kozbor, J. Immunol, 133:3001 (1984); Brodeur et ah, Monoclonal Antibody
- the hybridoma cells thus prepared are seeded and grown in a suitable culture medium, e.g., a medium that contains one or more substances that inhibit the growth or survival of the unfused, parental myeloma cells.
- a suitable culture medium e.g., a medium that contains one or more substances that inhibit the growth or survival of the unfused, parental myeloma cells.
- the culture medium for the hybridomas typically will include hypoxanthine, aminopterin, and thymidine (HAT medium), which substances prevent the growth of HGPRT-deficient cells.
- serum- free hybridoma cell culture methods are used to reduce use of animal-derived serum such as fetal bovine serum, as described, for example, in Even et al., Trends in Biotechnology, 24(3), 105-108 (2006).
- Oligopeptides as tools for improving productivity of hybridoma cell cultures are described in Franek, Trends in Monoclonal Antibody Research, 111-122 (2005).
- standard culture media are enriched with certain amino acids (alanine, serine, asparagine, proline), or with protein hydrolyzate fractions, and apoptosis may be significantly suppressed by synthetic oligopeptides, constituted of three to six amino acid residues.
- the peptides are present at millimolar or higher concentrations.
- Culture medium in which hybridoma cells are growing may be assayed for production of monoclonal antibodies that bind to mutant SMO.
- the binding specificity of monoclonal antibodies produced by hybridoma cells may be determined by immunoprecipitation or by an in vitro binding assay, such as radioimmunoassay (RIA) or enzyme-linked immunoadsorbent assay (ELISA).
- RIA radioimmunoassay
- ELISA enzyme-linked immunoadsorbent assay
- the binding affinity of the monoclonal antibody can be determined, for example, by Scatchard analysis. See, e.g., Munson et al, Anal. Biochem., 107:220 (1980).
- hybridoma cells After hybridoma cells are identified that produce antibodies of the desired specificity, affinity, and/or activity, the clones may be subcloned by limiting dilution procedures and grown by standard methods. See, e.g., Goding, supra. Suitable culture media for this purpose include, for example, D-MEM or RPMI-1640 medium.
- hybridoma cells may be grown in vivo as ascites tumors in an animal. Monoclonal antibodies secreted by the subclones are suitably separated from the culture medium, ascites fluid, or serum by conventional immunoglobulin purification procedures such as, for example, protein A-Sepharose, hydroxylapatite
- the method includes using minimal salts, such as lyotropic salts, in the binding process and preferably also using small amounts of organic solvents in the elution process.
- Antibodies of the invention can be made by using combinatorial libraries to screen for antibodies with the desired activity or activities.
- a variety of methods are known in the art for generating phage display libraries and screening such libraries for antibodies possessing the desired binding characteristics. Such methods are described generally in Hoogenboom et al. in Methods in Molecular Biology 178: 1-37 (O'Brien et al., ed., Human Press, Totowa, NJ, 2001).
- one method of generating antibodies of interest is through the use of a phage antibody library as described in Lee et al, J. Mol. Biol. (2004), 340(5): 1073-93.
- synthetic antibody clones are selected by screening phage libraries containing phage that display various fragments of antibody variable region (Fv) fused to phage coat protein. Such phage libraries are panned by affinity chromatography against the desired antigen. Clones expressing Fv fragments capable of binding to the desired antigen are adsorbed to the antigen and thus separated from the non-binding clones in the library. The binding clones are then eluted from the antigen, and can be further enriched by additional cycles of antigen
- Fv antibody variable region
- any of the antibodies of the invention can be obtained by designing a suitable antigen screening procedure to select for the phage clone of interest followed by construction of a full length antibody clone using the Fv sequences from the phage clone of interest and suitable constant region (Fc) sequences described in Kabat et al , Sequences of Proteins of Immunological Interest, Fifth Edition, NIH Publication 91-3242, Bethesda MD (1991), vols. 1-3.
- Fc constant region
- the antigen-binding domain of an antibody is formed from two variable (V) regions of about 110 amino acids, one each from the light (VL) and heavy (VH) chains, that both present three hypervariable loops (FJVRs) or complementarity-determining regions (CDRs).
- V variable
- VH variable
- CDRs complementarity-determining regions
- Variable domains can be displayed functionally on phage, either as single-chain Fv (scFv) fragments, in which VH and VL are covalently linked through a short, flexible peptide, or as Fab fragments, in which they are each fused to a constant domain and interact non-covalently, as described in Winter et ⁇ ., ⁇ . Rev. Immunol, 12: 433-455 (1994).
- scFv encoding phage clones and Fab encoding phage clones are collectively referred to as "Fv phage clones" or "Fv clones.”
- Repertoires of VH and VL genes can be separately cloned by polymerase chain reaction (PCR) and recombined randomly in phage libraries, which can then be searched for antigen-binding clones as described in Winter et al, Ann. Rev. Immunol, 12: 433-455 (1994).
- Libraries from immunized sources provide high-affinity antibodies to the immunogen without the requirement of constructing hybridomas.
- the naive repertoire can be cloned to provide a single source of human antibodies to a wide range of non-self and also self antigens without any
- naive libraries can also be made synthetically by cloning the unrearranged V-gene segments from stem cells, and using PCR primers containing random sequence to encode the highly variable CDR3 regions and to accomplish rearrangement in vitro as described by Hoogenboom and Winter, J. Mol. Biol, 227: 381-388 (1992).
- filamentous phage is used to display antibody fragments by fusion to the minor coat protein pill.
- the antibody fragments can be displayed as single chain Fv fragments, in which VH and VL domains are connected on the same polypeptide chain by a flexible polypeptide spacer, e.g. as described by Marks et al., J. Mol. Biol., 222: 581-597 (1991), or as Fab fragments, in which one chain is fused to pill and the other is secreted into the bacterial host cell periplasm where assembly of a Fab-coat protein structure which becomes displayed on the phage surface by displacing some of the wild type coat proteins, e.g. as described in Hoogenboom et al., Nucl. Acids Res., 19: 4133-4137 (1991).
- nucleic acids encoding antibody gene fragments are obtained from immune cells harvested from humans or animals. If a library biased in favor of anti- mutant SMO clones is desired, the subject is immunized with mutant SMO to generate an antibody response, and spleen cells and/or circulating B cells other peripheral blood lymphocytes (PBLs) are recovered for library construction.
- a human antibody gene fragment library biased in favor of anti-mutant SMO clones is obtained by generating an anti-mutant SMO antibody response in transgenic mice carrying a functional human immunoglobulin gene array (and lacking a functional endogenous antibody production system) such that mutant SMO immunization gives rise to B cells producing human antibodies against mutant SMO. The generation of human antibody-producing transgenic mice is described below.
- Additional enrichment for anti-mutant SMO reactive cell populations can be obtained by using a suitable screening procedure to isolate B cells expressing mutant SMO-specific membrane bound antibody, e.g., by cell separation using mutant SMO affinity chromatography or adsorption of cells to fluorochrome-labeled mutant SMO followed by flow-activated cell sorting (FACS).
- FACS flow-activated cell sorting
- spleen cells and/or B cells or other PBLs from an unimmunized donor provides a better representation of the possible antibody repertoire, and also permits the construction of an antibody library using any animal (human or non-human) species in which mutant SMO is not antigenic.
- stem cells are harvested from the subject to provide nucleic acids encoding unrearranged antibody gene segments.
- the immune cells of interest can be obtained from a variety of animal species, such as human, mouse, rat, lagomorpha, luprine, canine, feline, porcine, bovine, equine, and avian species, etc.
- Nucleic acid encoding antibody variable gene segments are recovered from the cells of interest and amplified.
- the desired DNA can be obtained by isolating genomic DNA or mRNA from lymphocytes followed by polymerase chain reaction (PCR) with primers matching the 5' and 3' ends of rearranged VH and VL genes as described in Orlandi et al., Proc. Natl. Acad. Sci. (USA), 86: 3833-3837 (1989), thereby making diverse V gene repertoires for expression.
- the V genes can be amplified from cDNA and genomic DNA, with back primers at the 5' end of the exon encoding the mature V-domain and forward primers based within the J-segment as described in Orlandi et al. (1989) and in Ward et al, Nature, 341 : 544-546 (1989).
- back primers can also be based in the leader exon as described in Jones et al, Biotechnol, 9: 88-89 (1991), and forward primers within the constant region as described in Sastry et al, Proc. Natl Acad. Sci. (USA), 86: 5728-5732 (1989).
- degeneracy can be used to maximize complementarity.
- library diversity is maximized by using PCR primers targeted to each V-gene family in order to amplify all available VH and VL arrangements present in the immune cell nucleic acid sample, e.g. as described in the method of Marks et al., J. Mol. Biol, 222: 581-597 (1991) or as described in the method of Oram et al, Nucleic Acids Res., 21 : 4491-4498 (1993).
- rare restriction sites can be introduced within the PCR primer as a tag at one end as described in Orlandi et al. (1989), or by further PCR amplification with a tagged primer as described in Clackson et al, Nature, 352: 624-628 (1991).
- Repertoires of synthetically rearranged V genes can be derived in vitro from V gene segments.
- Most of the human VH-gene segments have been cloned and sequenced (reported in Tomlinson et al., J. Mol. Biol, 227: 776-798 (1992)), and mapped (reported in Matsuda et al, Nature Genet., 3: 88-94 (1993); these cloned segments (including all the major conformations of the HI and H2 loop) can be used to generate diverse VH gene repertoires with PCR primers encoding H3 loops of diverse sequence and length as described in Hoogenboom and Winter, J. Mol. Biol, 227: 381-388 (1992).
- VH repertoires can also be made with all the sequence diversity focused in a long H3 loop of a single length as described in Barbas et al, Proc. Natl. Acad. Sci. USA, 89: 4457-4461 (1992).
- Human VK and ⁇ segments have been cloned and sequenced (reported in Williams and Winter, Eur. J. Immunol., 23: 1456- 1461 (1993)) and can be used to make synthetic light chain repertoires.
- Synthetic V gene repertoires based on a range of VH and VL folds, and L3 and H3 lengths, will encode antibodies of considerable structural diversity.
- germline V-gene segments can be rearranged in vitro according to the methods of Hoogenboom and Winter, J. Mol. Biol, 227: 381-388 (1992).
- Repertoires of antibody fragments can be constructed by combining VH and VL gene repertoires together in several ways. Each repertoire can be created in different vectors, and the vectors recombined in vitro, e.g., as described in Hogrefe et al, Gene, 128: 119-126 (1993), or in vivo by combinatorial infection, e.g., the loxP system described in Waterhouse et al. , Nucl. Acids Res., 21 : 2265-2266 (1993). The in vivo recombination approach exploits the two-chain nature of Fab fragments to overcome the limit on library size imposed by E. coli transformation efficiency.
- Naive VH and VL repertoires are cloned separately, one into a phagemid and the other into a phage vector.
- the two libraries are then combined by phage infection of phagemid-containing bacteria so that each cell contains a different combination and
- the library size is limited only by the number of cells present (about 10 clones). Both vectors contain in vivo recombination signals so that the VH and VL genes are recombined onto a single replicon and are co-packaged into phage virions. These huge libraries provide large numbers of diverse antibodies of good affinity (K 1 of about 10 "8 M).
- the repertoires may be cloned sequentially into the same vector, e.g. as described in Barbas et al., Proc. Natl. Acad. Sci. USA, 88: 7978-7982 (1991), or assembled together by PCR and then cloned, e.g. as described in Clackson et al. , Nature, 352: 624-628 (1991).
- PCR assembly can also be used to join VH and VL DNAs with DNA encoding a flexible peptide spacer to form single chain Fv (scFv) repertoires.
- in cell PCR assembly is used to combine VH and VL genes within lymphocytes by PCR and then clone repertoires of linked genes as described in Embleton et al., Nucl. Acids Res., 20: 3831-3837 (1992).
- the antibodies produced by naive libraries can be of moderate affinity (K ⁇ f 1 of about 10 6 to 10 7 M "1 ), but affinity maturation can also be mimicked in vitro by constructing and reselecting from secondary libraries as described in Winter et al. (1994), supra.
- mutation can be introduced at random in vitro by using error-prone polymerase (reported in Leung et al.
- affinity maturation can be performed by randomly mutating one or more CDRs, e.g. using PCR with primers carrying random sequence spanning the CDR of interest, in selected individual Fv clones and screening for higher affinity clones.
- WO 9607754 published 14 March 1996) described a method for inducing mutagenesis in a complementarity determining region of an immunoglobulin light chain to create a library of light chain genes.
- VH or VL domains selected by phage display with repertoires of naturally occurring V domain variants obtained from unimmunized donors and screen for higher affinity in several rounds of chain reshuffling as described in Marks et al, Biotechnol, 10: 779- 783 (1992).
- This technique allows the production of antibodies and antibody fragments with affinities of about 10 "9 M or less.
- mutant SMO can be used to coat the wells of adsorption plates, expressed on host cells affixed to adsorption plates or used in cell sorting, or conjugated to biotin for capture with streptavidin-coated beads, or used in any other method for panning phage display libraries.
- the phage library samples are contacted with immobilized mutant SMO under conditions suitable for binding at least a portion of the phage particles with the adsorbent. Normally, the conditions, including pH, ionic strength, temperature and the like are selected to mimic physiological conditions.
- the phages bound to the solid phase are washed and then eluted by acid, e.g. as described in Barbas et al, Proc. Natl. Acad. Sci USA, 88: 7978-7982 (1991), or by alkali, e.g. as described in Marks et al. , J. Mol. Biol, 222: 581-597 (1991), or by mutant SMO antigen competition, e.g.
- Phages can be enriched 20-1,000-fold in a single round of selection. Moreover, the enriched phages can be grown in bacterial culture and subjected to further rounds of selection. The efficiency of selection depends on many factors, including the kinetics of dissociation during washing, and whether multiple antibody fragments on a single phage can simultaneously engage with antigen. Antibodies with fast dissociation kinetics (and weak binding affinities) can be retained by use of short washes, multivalent phage display and high coating density of antigen in solid phase.
- the high density not only stabilizes the phage through multivalent interactions, but favors rebinding of phage that has dissociated.
- the selection of antibodies with slow dissociation kinetics (and good binding affinities) can be promoted by use of long washes and monovalent phage display as described in Bass et ah, Proteins, 8: 309- 314 (1990) and in WO 92/09690, and a low coating density of antigen as described in Marks et al, Biotechnol, 10: 779-783 (1992).
- phage antibodies of different affinities can be selected between phage antibodies of different affinities, even with affinities that differ slightly, for mutant SMO.
- random mutation of a selected antibody ⁇ e.g. as performed in some affinity maturation techniques) is likely to give rise to many mutants, most binding to antigen, and a few with higher affinity.
- rare high affinity phage could be competed out.
- phages can be incubated with excess biotinylated mutant SMO, but with the biotinylated mutant SMO at a concentration of lower molarity than the target molar affinity constant for mutant SMO.
- the high affinity-binding phages can then be captured by streptavidin-coated paramagnetic beads.
- Anti-mutant SMO clones may be selected based on activity.
- the invention provides anti-mutant SMO antibodies that bind to living cells that naturally express mutant SMO, such as GDC-0449-resistant tumor cells.
- the invention provides anti-mutant SMO antibodies that bind to the same region as that bound by GDC-0449 in wild type SMO.
- Fv clones corresponding to such anti -mutant SMO antibodies can be selected by (1) isolating anti-mutant SMO clones from a phage library as described above, and optionally amplifying the isolated population of phage clones by growing up the population in a suitable bacterial host; (2) selecting mutant SMO and a second protein against which blocking and non- blocking activity, respectively, is desired; (3) adsorbing the anti-mutant SMO phage clones to immobilized mutant SMO; (4) using an excess of the second protein to elute any undesired clones that recognize mutant SMO-binding determinants which overlap or are shared with the binding determinants of the second protein; and (5) eluting the clones which remain adsorbed following step (4).
- clones with the desired blocking/non-blocking properties can be further enriched by repeating the selection procedures described herein one or more times.
- DNA encoding hybridoma-derived monoclonal antibodies or phage display Fv clones of the invention is readily isolated and sequenced using conventional procedures (e.g. by using oligonucleotide primers designed to specifically amplify the heavy and light chain coding regions of interest from hybridoma or phage DNA template).
- the DNA can be placed into expression vectors, which are then transfected into host cells such as E. coli cells, simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of the desired monoclonal antibodies in the recombinant host cells.
- DNA encoding the Fv clones of the invention can be combined with known DNA sequences encoding heavy chain and/or light chain constant regions (e.g. the appropriate DNA sequences can be obtained from Kabat et ah, supra) to form clones encoding full or partial length heavy and/or light chains.
- constant regions of any isotype can be used for this purpose, including IgG, IgM, IgA, IgD, and IgE constant regions, and that such constant regions can be obtained from any human or animal species.
- an Fv clone derived from the variable domain DNA of one animal (such as human) species and then fused to constant region DNA of another animal species to form coding sequence(s) for "hybrid," full length heavy chain and/or light chain is included in the definition of "chimeric” and "hybrid” antibody as used herein.
- an Fv clone derived from human variable DNA is fused to human constant region DNA to form coding sequence(s) for full- or partial-length human heavy and/or light chains.
- DNA encoding anti-mutant SMO antibody derived from a hybridoma of the invention can also be modified, for example, by substituting the coding sequence for human heavy- and light-chain constant domains in place of homologous murine sequences derived from the hybridoma clone (e.g. as in the method of Morrison et al., Proc. Natl. Acad. Sci. USA, 81 : 6851-6855 (1984)).
- DNA encoding a hybridoma- or Fv clone-derived antibody or fragment can be further modified by covalently joining to the immunoglobulin coding sequence all or part of the coding sequence for a non- immunoglobulin polypeptide. In this manner, "chimeric" or “hybrid” antibodies are prepared that have the binding specificity of the Fv clone or hybridoma clone-derived antibodies of the invention.
- Antibodies may also be produced using recombinant methods.
- nucleic acid encoding the antibody is isolated and inserted into a replicable vector for further cloning
- DNA encoding the antibody may be readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of the antibody).
- Many vectors are available.
- the vector components generally include, but are not limited to, one or more of the following: a signal sequence, an origin of replication, one or more marker genes, an enhancer element, a promoter, and a transcription termination sequence.
- An antibody of the invention may be produced recombinantly not only directly, but also as a fusion polypeptide with a heterologous polypeptide, which is preferably a signal sequence or other polypeptide having a specific cleavage site at the N-terminus of the mature protein or polypeptide.
- a heterologous polypeptide which is preferably a signal sequence or other polypeptide having a specific cleavage site at the N-terminus of the mature protein or polypeptide.
- the heterologous signal sequence selected preferably is one that is recognized and processed (i.e., cleaved by a signal peptidase) by the host cell.
- the signal sequence is substituted by a prokaryotic signal sequence selected, for example, from the group of the alkaline phosphatase, penicillinase, lpp, or heat-stable enterotoxin II leaders.
- a prokaryotic signal sequence selected, for example, from the group of the alkaline phosphatase, penicillinase, lpp, or heat-stable enterotoxin II leaders.
- yeast secretion the native signal sequence may be substituted by, e.g., the yeast invertase leader, a factor leader (including Saccharomyces and Kluyveromyces a-factor leaders), or acid phosphatase leader, the C. albicans glucoamylase leader, or the signal described in WO 90/13646.
- mammalian signal sequences as well as viral secretory leaders for example, the herpes simplex gD signal, are available.
- Both expression and cloning vectors contain a nucleic acid sequence that enables the vector to replicate in one or more selected host cells.
- this sequence is one that enables the vector to replicate independently of the host chromosomal DNA, and includes origins of replication or autonomously replicating sequences.
- origins of replication or autonomously replicating sequences are well known for a variety of bacteria, yeast, and viruses.
- the origin of replication from the plasmid pBR322 is suitable for most Gram-negative bacteria, the 2 ⁇ plasmid origin is suitable for yeast, and various viral origins (SV40, polyoma, adenovirus, VSV or BPV) are useful for cloning vectors in mammalian cells.
- the origin of replication component is not needed for mammalian expression vectors (the SV40 origin may typically be used only because it contains the early promoter).
- Selection genes may contain a selection gene, also termed a selectable marker.
- Typical selection genes encode proteins that (a) confer resistance to antibiotics or other toxins, e.g., ampicillin, neomycin, methotrexate, or tetracycline, (b) complement auxotrophic deficiencies, or (c) supply critical nutrients not available from complex media, e.g., the gene encoding D-alanine racemase for Bacilli.
- One example of a selection scheme utilizes a drug to arrest growth of a host cell. Those cells that are successfully transformed with a heterologous gene produce a protein conferring drug resistance and thus survive the selection regimen. Examples of such dominant selection use the drugs neomycin, mycophenolic acid and hygromycin.
- Suitable selectable markers for mammalian cells are those that enable the identification of cells competent to take up antibody-encoding nucleic acid, such as DHFR, glutamine synthetase (GS), thymidine kinase, metallothionein-I and -II, preferably primate metallothionein genes, adenosine deaminase, ornithine decarboxylase, etc.
- cells transformed with the DHFR gene are identified by culturing the transformants in a culture medium containing methotrexate (Mtx), a competitive antagonist of DHFR. Under these conditions, the DHFR gene is amplified along with any other co-transformed nucleic acid.
- Mtx methotrexate
- a Chinese hamster ovary (CHO) cell line deficient in endogenous DHFR activity e.g., ATCC CRL- 9096 may be used.
- cells transformed with the GS gene are identified by culturing the transformants in a culture medium containing L-methionine sulfoximine (Msx), an inhibitor of GS. Under these conditions, the GS gene is amplified along with any other co-transformed nucleic acid.
- the GS selection/amplification system may be used in combination with the DHFR selection/amplification system described above.
- host cells transformed or co-transformed with DNA sequences encoding an antibody of interest, wild-type DHFR gene, and another selectable marker such as aminoglycoside 3 '-phosphotransferase (APH) can be selected by cell growth in medium containing a selection agent for the selectable marker such as an aminoglycosidic antibiotic, e.g., kanamycin, neomycin, or G418. See U.S. Patent No. 4,965,199.
- APH aminoglycoside 3 '-phosphotransferase
- a suitable selection gene for use in yeast is the trpl gene present in the yeast plasmid YRp7 (Stinchcomb et ah, Nature, 282:39 (1979)).
- the trpl gene provides a selection marker for a mutant strain of yeast lacking the ability to grow in tryptophan, for example, ATCC No. 44076 or PEP4-1. Jones, Genetics, 85: 12 (1977).
- the presence of the trpl lesion in the yeast host cell genome then provides an effective environment for detecting transformation by growth in the absence of tryptophan.
- ZeM2-deficient yeast strains (ATCC 20,622 or 38,626) are complemented by known plasmids bearing the Leul gene.
- vectors derived from the 1.6 ⁇ circular plasmid pKDl can be used for transformation of Kluyveromyces yeasts.
- an expression system for large-scale production of recombinant calf chymosin was reported for K. lactis. Van den Berg, Bio/Technology, 8: 135 (1990).
- Stable multi-copy expression vectors for secretion of mature recombinant human serum albumin by industrial strains of Kluyveromyces have also been disclosed. Fleer et ah, Bio/Technology, 9:968-975 (1991).
- Expression and cloning vectors generally contain a promoter that is recognized by the host organism and is operably linked to nucleic acid encoding an antibody.
- Promoters suitable for use with prokaryotic hosts include the phoA promoter , ⁇ - lactamase and lactose promoter systems, alkaline phosphatase promoter, a tryptophan (trp) promoter system, and hybrid promoters such as the tac promoter.
- trp tryptophan
- Other known bacterial promoters are suitable. Promoters for use in bacterial systems also will contain a Shine-Dalgarno (S.D.) sequence operably linked to the DNA encoding an antibody.
- S.D. Shine-Dalgarno
- Promoter sequences are known for eukaryotes. Virtually all eukaryotic genes have an AT -rich region located approximately 25 to 30 bases upstream from the site where transcription is initiated. Another sequence found 70 to 80 bases upstream from the start of transcription of many genes is a CNCAAT region where N may be any nucleotide. At the 3' end of most eukaryotic genes is an AATAAA sequence that may be the signal for addition of the poly A tail to the 3' end of the coding sequence. All of these sequences are suitably inserted into eukaryotic expression vectors.
- suitable promoter sequences for use with yeast hosts include the promoters for 3-phosphoglycerate kinase or other glycolytic enzymes, such as enolase, glyceraldehyde-3 -phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phosphofructokinase, glucose-6-phosphate isomerase, 3- phosphoglycerate mutase, pyruvate kinase, triosephosphate isomerase,
- yeast promoters which are inducible promoters having the additional advantage of transcription controlled by growth conditions, are the promoter regions for alcohol dehydrogenase 2, isocytochrome C, acid phosphatase, degradative enzymes associated with nitrogen metabolism, metallothionein, glyceraldehyde-3 - phosphate dehydrogenase, and enzymes responsible for maltose and galactose utilization.
- Suitable vectors and promoters for use in yeast expression are further described in EP 73,657.
- Yeast enhancers also are advantageously used with yeast promoters.
- Antibody transcription from vectors in mammalian host cells can be controlled, for example, by promoters obtained from the genomes of viruses such as polyoma virus, fowlpox virus, adenovirus (such as Adenovirus 2), bovine papilloma virus, avian sarcoma virus, cytomegalovirus, a retrovirus, hepatitis-B virus, Simian Virus 40 (SV40), or from heterologous mammalian promoters, e.g., the actin promoter or an immunoglobulin promoter, from heat-shock promoters, provided such promoters are compatible with the host cell systems.
- viruses such as polyoma virus, fowlpox virus, adenovirus (such as Adenovirus 2), bovine papilloma virus, avian sarcoma virus, cytomegalovirus, a retrovirus, hepatitis-B virus, Simian Virus 40 (SV40), or from hetero
- the early and late promoters of the SV40 virus are conveniently obtained as an SV40 restriction fragment that also contains the SV40 viral origin of replication.
- the immediate early promoter of the human cytomegalovirus is conveniently obtained as a Hindlll E restriction fragment.
- a system for expressing DNA in mammalian hosts using the bovine papilloma virus as a vector is disclosed in U.S. Patent No. 4,419,446. A modification of this system is described in U.S. Patent No. 4,601,978. See also Reyes et al., Nature 297:598-601 (1982) on expression of human ⁇ -interferon cDNA in mouse cells under the control of a thymidine kinase promoter from herpes simplex virus. Alternatively, the Rous Sarcoma Virus long terminal repeat can be used as the promoter.
- Enhancer sequences are now known from mammalian genes (globin, elastase, albumin, a-fetoprotein, and insulin). Typically, however, one will use an enhancer from a eukaryotic cell virus. Examples include the SV40 enhancer on the late side of the replication origin (bp 100-270), the cytomegalovirus early promoter enhancer, the polyoma enhancer on the late side of the replication origin, and adenovirus enhancers. See also Yaniv, Nature 297: 17-18 (1982) on enhancing elements for activation of eukaryotic promoters. The enhancer may be spliced into the vector at a position 5' or 3' to the antibody-encoding sequence, but is preferably located at a site 5' from the promoter.
- Expression vectors used in eukaryotic host cells will also contain sequences necessary for the termination of transcription and for stabilizing the mRNA. Such sequences are commonly available from the 5' and, occasionally 3', untranslated regions of eukaryotic or viral DNAs or cDNAs. These regions contain nucleotide segments transcribed as polyadenylated fragments in the untranslated portion of the mRNA encoding antibody.
- One useful transcription termination component is the bovine growth hormone polyadenylation region. See W094/11026 and the expression vector disclosed therein.
- Suitable host cells for cloning or expressing the DNA in the vectors herein are the prokaryote, yeast, or higher eukaryote cells described above.
- Suitable prokaryotes for this purpose include eubacteria, such as Gram-negative or Gram-positive organisms, for example, Enterobacteriaceae such as Escherichia, e.g., E. coli, Enterobacter, Erwinia, Klebsiella, Proteus, Salmonella, e.g., Salmonella
- E. coli cloning host is E. coli 294 (ATCC 31,446), although other strains such as E. coli B, E. coli XI 776 (ATCC 31,537), and E. coli W3110 (ATCC 27,325) are suitable. These examples are illustrative rather than limiting.
- Full length antibody, antibody fusion proteins, and antibody fragments can be produced in bacteria, in particular when glycosylation and Fc effector function are not needed, such as when the therapeutic antibody is conjugated to a cytotoxic agent ⁇ e.g., a toxin) that by itself shows effectiveness in tumor cell destruction.
- Full length antibodies have greater half life in circulation. Production in E. coli is faster and more cost efficient.
- cytotoxic agent e.g., a toxin
- the antibody may be isolated from the E. coli cell paste in a soluble fraction and can be purified through, e.g., a protein A or G column depending on the isotype. Final purification can be carried out similar to the process for purifying antibody expressed e.g,, in CHO cells.
- eukaryotic microbes such as filamentous fungi or yeast are suitable cloning or expression hosts for antibody-encoding vectors.
- Saccharomyces cerevisiae or common baker's yeast, is the most commonly used among lower eukaryotic host microorganisms. However, a number of other genera, species, and strains are commonly available and useful herein, such as
- Schizosaccharomyces pombe Kluyveromyces hosts such as, e.g., K. lactis, K.fragilis (ATCC 12,424), K. bulgaricus (ATCC 16,045), K. wickeramii (ATCC 24,178), K. waltii (ATCC 56,500), K. drosophilarum (ATCC 36,906), K . thermotolerans, and K. marxianus; yarrowia (EP 402,226); Pichia pastoris (EP 183,070); Candida;
- Trichoderma reesia (EP 244,234); Neurospora crassa; Schwanniomyces such as Schwanniomyces occidentalis; and filamentous fungi such as, e.g., Neurospora, Penicillium, Tolypocladium, and Aspergillus hosts such as A. nidulans and A. niger.
- filamentous fungi such as, e.g., Neurospora, Penicillium, Tolypocladium, and Aspergillus hosts such as A. nidulans and A. niger.
- Certain fungi and yeast strains may be selected in which glycosylation pathways have been "humanized,” resulting in the production of an antibody with a partially or fully human glycosylation pattern. See, e.g. , Li et al. , Nat. Biotech.
- Suitable host cells for the expression of glycosylated antibody are also derived from multicellular organisms (invertebrates and vertebrates). Examples of invertebrate cells include plant and insect cells. Numerous baculoviral strains and variants and corresponding permissive insect host cells from hosts such as Spodoptera frugiperda (caterpillar), Aedes aegypti (mosquito), Aedes albopictus (mosquito), Drosophila melanogaster (fruitfly), and Bombyx mori have been identified. A variety of viral strains for transfection are publicly available, e.g., the L-l variant of
- Autographa calif ornica NPV and the Bm-5 strain of Bombyx mori NPV, and such viruses may be used as the virus herein according to the present invention, particularly for transfection of Spodoptera frugiperda cells.
- Vertebrate cells may be used as hosts, and propagation of vertebrate cells in culture (tissue culture) has become a routine procedure.
- useful mammalian host cell lines are monkey kidney CV1 line transformed by SV40 (COS- 7, ATCC CRL 1651); human embryonic kidney line (293 or 293 cells subcloned for growth in suspension culture, Graham et al, J. Gen Virol. 36:59 (1977)) ; baby hamster kidney cells (BHK, ATCC CCL 10); mouse Sertoli cells (TM4, Mather, Biol. Reprod.
- monkey kidney cells (CV1 ATCC CCL 70); African green monkey kidney cells (VERO-76, ATCC CRL-1587); human cervical carcinoma cells (HELA, ATCC CCL 2); canine kidney cells (MDCK, ATCC CCL 34); buffalo rat liver cells (BRL 3A, ATCC CRL 1442); human lung cells (W138, ATCC CCL 75); human liver cells (Hep G2, HB 8065); mouse mammary tumor (MMT 060562, ATCC CCL51); TRI cells (Mather et al, Annals N. Y. Acad. Sci. 383:44-68 (1982)); MRC 5 cells; FS4 cells; and a human hepatoma line (Hep G2).
- CHO Chinese hamster ovary
- DHFR " CHO cells (Urlaub et al, Proc. Natl. Acad. Sci. USA 77:4216 (1980)); and myeloma cell lines such as NS0 and Sp2/0.
- CHO Chinese hamster ovary
- myeloma cell lines such as NS0 and Sp2/0.
- Host cells are transformed with the above-described expression or cloning vectors for antibody production and cultured in conventional nutrient media modified as appropriate for inducing promoters, selecting transformants, or amplifying the genes encoding the desired sequences.
- the host cells used to produce an antibody of this invention may be cultured in a variety of media.
- Commercially available media such as Ham's F10 (Sigma), Minimal Essential Medium ((MEM), (Sigma), RPMI-1640 (Sigma), and Dulbecco's Modified Eagle's Medium ((DMEM), Sigma) are suitable for culturing the host cells.
- any of these media may be supplemented as necessary with hormones and/or other growth factors (such as insulin, transferrin, or epidermal growth factor), salts (such as sodium chloride, calcium, magnesium, and phosphate), buffers (such as HEPES), nucleotides (such as adenosine and thymidine), antibiotics (such as GENTAMYCINTM drug), trace elements (defined as inorganic compounds usually present at final concentrations in the micromolar range), and glucose or an equivalent energy source. Any other necessary supplements may also be included at appropriate concentrations that would be known to those skilled in the art.
- the culture conditions such as temperature, pH, and the like, are those previously used with the host cell selected for expression, and will be apparent to the ordinarily skilled artisan.
- the antibody When using recombinant techniques, the antibody can be produced
- the particulate debris either host cells or lysed fragments, are removed, for example, by centrifugation or ultrafiltration.
- Carter et al., Bio/Technology 10: 163-167 (1992) describe a procedure for isolating antibodies which are secreted to the periplasmic space of E. coli. Briefly, cell paste is thawed in the presence of sodium acetate (pH 3.5), EDTA, and
- phenylmethylsulfonylfluoride over about 30 min. Cell debris can be removed by centrifugation.
- supematants from such expression systems are generally first concentrated using a commercially available protein concentration filter, for example, an Amicon or Millipore Pellicon ultrafiltration unit.
- a protease inhibitor such as PMSF may be included in any of the foregoing steps to inhibit proteolysis and antibiotics may be included to prevent the growth of adventitious contaminants.
- the antibody composition prepared from the cells can be purified using, for example, hydroxylapatite chromatography, hydrophobic interaction chromatography, gel electrophoresis, dialysis, and affinity chromatography, with affinity
- protein A as an affinity ligand depends on the species and isotype of any immunoglobulin Fc domain that is present in the antibody. Protein A can be used to purify antibodies that are based on human ⁇ , ⁇ 2, or ⁇ 4 heavy chains (Lindmark et al., J. Immunol. Meth. 62: 1-13 (1983)). Protein G is recommended for all mouse isotypes and for human ⁇ 3 (Guss et al., EMBO J. 5: 15671575 (1986)). The matrix to which the affinity ligand is attached is most often agarose, but other matrices are available. Mechanically stable matrices such as controlled pore glass or
- poly(styrenedivinyl)benzene allow for faster flow rates and shorter processing times than can be achieved with agarose.
- the Bakerbond ABXTMresin J. T. Baker, Phillipsburg, NJ
- Other techniques for protein purification such as fractionation on an ion-exchange column, ethanol precipitation, Reverse Phase HPLC, chromatography on silica, chromatography on heparin SEPHAROSETM chromatography on an anion or cation exchange resin (such as a polyaspartic acid column), chromatofocusing, SDS-PAGE, and ammonium sulfate precipitation are also available depending on the antibody to be recovered.
- the mixture comprising the antibody of interest and contaminants may be subjected to low pH hydrophobic interaction chromatography using an elution buffer at a pH between about 2.5-4.5, preferably performed at low salt concentrations (e.g., from about 0-0.25M salt).
- the invention also provides immunoconjugates (interchangeably referred to as "antibody-drug conjugates,” or “ADCs”) comprising an antibody conjugated to one or more cytotoxic agents, such as a chemotherapeutic agent, a drug, a growth inhibitory agent, a toxin (e.g., a protein toxin, an enzymatically active toxin of bacterial, fungal, plant, or animal origin, or fragments thereof), or a radioactive isotope (i.e., a radioconjugate).
- cytotoxic agents such as a chemotherapeutic agent, a drug, a growth inhibitory agent, a toxin (e.g., a protein toxin, an enzymatically active toxin of bacterial, fungal, plant, or animal origin, or fragments thereof), or a radioactive isotope (i.e., a radioconjugate).
- Immunoconjugates have been used for the local delivery of cytotoxic agents, i.e., drugs that kill or inhibit the growth or proliferation of cells, in the treatment of cancer (Lambert, J. (2005) Curr. Opinion in Pharmacology 5:543-549; Wu et al
- Immunoconjugates allow for the targeted delivery of a drug moiety to a tumor, and intracellular accumulation therein, where systemic administration of unconjugated drugs may result in unacceptable levels of toxicity to normal cells as well as the tumor cells sought to be eliminated (Baldwin et al, Lancet (Mar. 15, 1986) pp. 603-05; Thorpe (1985) "Antibody Carriers Of Cytotoxic Agents In Cancer Therapy: A
- geldanamycin (Mandler et al (2000) J. Nat. Cancer Inst. 92(19): 1573-1581; Mandler et al (2000) Bioorganic & Med. Chem. Letters 10: 1025-1028; Mandler et al (2002) Bioconjugate Chem. 13:786-791), maytansinoids (EP 1391213; Liu et al, (1996) Proc. Natl. Acad. Sci. USA 93:8618-8623), and calicheamicin (Lode et al (1998) Cancer Res. 58:2928; Hinman et al (1993) Cancer Res. 53:3336-3342).
- the toxins may exert their cytotoxic effects by mechanisms including tubulin binding, DNA binding, or topoisomerase inhibition. Some cytotoxic drugs tend to be inactive or less active when conjugated to large antibodies or protein receptor ligands.
- ZEVALIN® is an antibody-radioisotope conjugate composed of a murine IgGl kappa monoclonal antibody directed against the CD20 antigen found on the surface of normal and malignant B lymphocytes and 11 lln or 90 Y radioisotope bound by a thiourea linker-chelator (Wiseman et al (2000) Eur. Jour. Nucl. Med. 27(7):766-77; Wiseman et al (2002) Blood 99(12):4336-42; Witzig et al (2002) J. Clin. Oncol.
- ZEVALIN has activity against B-cell non-Hodgkin's Lymphoma (NHL), administration results in severe and prolonged cytopenias in most patients.
- MYLOTARGTM (gemtuzumab ozogamicin, Wyeth Pharmaceuticals), an antibody-drug conjugate composed of a huCD33 antibody linked to calicheamicin, was approved in 2000 for the treatment of acute myeloid leukemia by injection
- an antibody-drug conjugate composed of the huC242 antibody linked via the disulfide linker SPP to the maytansinoid drug moiety, DM1 is advancing into Phase II trials for the treatment of cancers that express CanAg, such as colon, pancreatic, gastric, and other cancers.
- MLN-2704 (Millennium Pharm., BZL Biologies, Immunogen Inc.), an antibody-drug conjugate composed of the anti- prostate specific membrane antigen (PSMA) monoclonal antibody linked to the maytansinoid drug moiety, DM1, is under development for the potential treatment of prostate tumors.
- PSMA prostate specific membrane antigen
- auristatin peptides auristatin E (AE) and monomethylauristatin (MMAE), synthetic analogs of dolastatin, were conjugated to chimeric monoclonal antibodies cBR96 (specific to Lewis Y on carcinomas) and cACIO (specific to CD30 on hematological malignancies) (Doronina et al (2003) Nature Biotechnol. 21(7):778- 784) and are under therapeutic development.
- an immunoconjugate comprises an antibody and a chemotherapeutic agent or other toxin.
- Chemotherapeutic agents useful in the generation of immunoconjugates are described herein ⁇ e.g., above).
- Enzymatically active toxins and fragments thereof that can be used include diphtheria A chain, nonbinding active fragments of diphtheria toxin, exotoxin A chain (from
- Pseudomonas aeruginosa Pseudomonas aeruginosa
- ricin A chain abrin A chain, modeccin A chain, alpha- sarcin, Aleurites fordii proteins, dianthin proteins, Phytolaca americana proteins (PAPI, PAP II, and PAP-S), momordica charantia inhibitor, curcin, crotin, sapaonaria officinalis inhibitor, gelonin, mitogellin, restrictocin, phenomycin, enomycin, and the tricothecenes. See, e.g., WO 93/21232 published October 28, 1993.
- a variety of radionuclides are available for the production of radioconjugated antibodies.
- Conjugates of the antibody and cytotoxic agent are made using a variety of bifunctional protein-coupling agents such as N-succinimidyl-3-(2-pyridyldithiol) propionate (SPDP), iminothiolane (IT), bifunctional derivatives of imidoesters (such as dimethyl adipimidate HC1), active esters (such as disuccinimidyl suberate), aldehydes (such as glutaraldehyde), bis-azido compounds (such as bis (p-azidobenzoyl) hexanediamine), bis-diazonium derivatives (such as bis-(p-diazoniumbenzoyl)-ethylenediamine), diisocyanates (such as toluene 2,6-diisocyanate), and bis-active fluorine compounds (such as l,5-difluoro
- SPDP N-succinimidyl-3-(2-pyridyl
- a ricin immunotoxin can be prepared as described in Vitetta et al., Science, 238: 1098 (1987).
- Carbon- 14-labeled 1-isothiocyanatobenzyl- 3-methyldiethylene triaminepentaacetic acid (MX-DTPA) is an exemplary chelating agent for conjugation of radionucleotide to the antibody. See W094/11026.
- Conjugates of an antibody and one or more small molecule toxins such as a calicheamicin, maytansinoids, dolastatins, aurostatins, a trichothecene, and CC1065, and the derivatives of these toxins that have toxin activity, are also contemplated herein.
- the immunoconjugate comprises an antibody (full length or fragments) conjugated to one or more maytansinoid molecules.
- Maytansinoids are mitototic inhibitors which act by inhibiting tubulin polymerization. Maytansine was first isolated from the east African shrub Maytenus serrata (U.S. Patent No. 3,896,111). Subsequently, it was discovered that certain microbes also produce maytansinoids, such as maytansinol and C-3 maytansinol esters (U.S. Patent No. 4,151,042). Synthetic maytansinol and derivatives and analogues thereof are disclosed, for example, in U.S. Patent Nos. 4,137,230;
- Maytansinoid drug moieties are attractive drug moieties in antibody drug conjugates because they are: (i) relatively accessible to prepare by fermentation or chemical modification, derivatization of fermentation products, (ii) amenable to derivatization with functional groups suitable for conjugation through the non- disulfide linkers to antibodies, (iii) stable in plasma, and (iv) effective against a variety of tumor cell lines.
- the drug conjugate achieved a degree of cytotoxicity similar to the free maytansinoid drug, which could be increased by increasing the number of maytansinoid molecules per antibody molecule.
- the A7 -maytansinoid conjugate showed low systemic cytotoxicity in mice.
- Antibody-maytansinoid conjugates are prepared by chemically linking an antibody to a maytansinoid molecule without significantly diminishing the biological activity of either the antibody or the maytansinoid molecule. See, e.g., U.S. Patent No. 5,208,020 (the disclosure of which is hereby expressly incorporated by reference). An average of 3-4 maytansinoid molecules conjugated per antibody molecule has shown efficacy in enhancing cytotoxicity of target cells without negatively affecting the function or solubility of the antibody, although even one molecule of toxin/antibody would be expected to enhance cytotoxicity over the use of naked antibody. Maytansinoids are well known in the art and can be synthesized by known techniques or isolated from natural sources.
- Suitable maytansinoids are disclosed, for example, in U.S. Patent No. 5,208,020 and in the other patents and nonpatent publications referred to hereinabove.
- Preferred maytansinoids are maytansinol and maytansinol analogues modified in the aromatic ring or at other positions of the maytansinol molecule, such as various maytansinol esters.
- Antibody- maytansinoid conjugates comprising the linker component SMCC may be prepared as disclosed in U.S. Patent Application No. 10/960,602, filed Oct. 8, 2004.
- the linking groups include disulfide groups, thioether groups, acid labile groups, photolabile groups, peptidase labile groups, or esterase labile groups, as disclosed in the above- identified patents, disulfide and thioether groups being preferred. Additional linking groups are described and exemplified herein.
- Conjugates of the antibody and maytansinoid may be made using a variety of bifunctional protein coupling agents such as N-succinimidyl-3-(2-pyridyldithio) propionate (SPDP), succinimidyl-4-(N-maleimidomethyl) cyclohexane-l-carboxylate (SMCC), iminothiolane (IT), bifunctional derivatives of imidoesters (such as dimethyl adipimidate HC1), active esters (such as disuccinimidyl suberate), aldehydes (such as glutaraldehyde), bis-azido compounds (such as bis (p-azidobenzoyl) hexanediamine), bis-diazonium derivatives (such as bis-(p-diazoniumbenzoyl)-ethylenediamine), diisocyanates (such as toluene 2,6-diisocyanate), and bis-active fluorine compounds
- Particularly preferred coupling agents include N-succinimidyl-3-(2-pyridyldithio) propionate (SPDP) (Carlsson et al, Biochem. J. 173:723-737 (1978)) and N-succinimidyl-4-(2-pyridylthio)pentanoate (SPP) to provide for a disulfide linkage.
- SPDP N-succinimidyl-3-(2-pyridyldithio) propionate
- SPP N-succinimidyl-4-(2-pyridylthio)pentanoate
- the linker may be attached to the maytansinoid molecule at various positions, depending on the type of the link. For example, an ester linkage may be formed by reaction with a hydroxyl group using conventional coupling techniques.
- the reaction may occur at the C-3 position having a hydroxyl group, the C-14 position modified with hydroxymethyl, the C-15 position modified with a hydroxyl group, and the C-20 position having a hydroxyl group.
- the linkage is formed at the C-3 position of maytansinol or a maytansinol analogue.
- the dolastatin or auristatin drug moiety may be attached to the antibody through the N (amino) terminus or the C (carboxyl) terminus of the peptidic drug moiety (WO 02/088172).
- the immunoconjugate comprises an antibody conjugated to one or more calicheamicin molecules.
- the calicheamicin family of antibiotics are capable of producing double-stranded DNA breaks at sub-picomolar concentrations.
- For the preparation of conjugates of the calicheamicin family see U.S. patents 5,712,374, 5,714,586, 5,739,116, 5,767,285, 5,770,701, 5,770,710, 5,773,001, 5,877,296 (all to American Cyanamid Company).
- Structural analogues of calicheamicin which may be used include, but are not limited to, ⁇ , ⁇ 2 ⁇ , ⁇ 3 ⁇ , N- acetyl- ⁇ , PSAG and ⁇ 1 (Hinman et al, Cancer Research 53:3336-3342 (1993), Lode et al., Cancer Research 58:2925-2928 (1998) and the aforementioned U.S. patents to American Cyanamid).
- Another anti-tumor drug that the antibody can be conjugated is QFA which is an antifolate.
- QFA is an antifolate.
- Both calicheamicin and QFA have intracellular sites of action and do not readily cross the plasma membrane. Therefore, cellular uptake of these agents through antibody mediated internalization greatly enhances their cytotoxic effects.
- antitumor agents that can be conjugated to the antibodies include BCNU, streptozoicin, vincristine and 5-fluorouracil, the family of agents known collectively LL-E33288 complex described in U.S. patents 5,053,394, 5,770,710, as well as esperamicins (U.S. patent 5,877,296).
- Enzymatically active toxins and fragments thereof which can be used include diphtheria A chain, nonbinding active fragments of diphtheria toxin, exotoxin A chain (from Pseudomonas aeruginosa), ricin A chain, abrin A chain, modeccin A chain, alpha-sarcin, Aleurites fordii proteins, dianthin proteins, Phytolaca americana proteins (PAPI, PAP II, and PAP-S), momordica charantia inhibitor, curcin, crotin, sapaonaria officinalis inhibitor, gelonin, mitogellin, restrictocin, phenomycin, enomycin and the tricothecenes. See, for example, WO 93/21232 published October 28, 1993.
- the present invention further contemplates an immunoconjugate formed between an antibody and a compound with nucleolytic activity ⁇ e.g. , a ribonuclease or a DNA endonuclease such as a deoxyribonuclease; DNase).
- a compound with nucleolytic activity e.g. , a ribonuclease or a DNA endonuclease such as a deoxyribonuclease; DNase).
- the antibody may comprise a highly radioactive atom.
- a variety of radioactive isotopes are available for the production of radioconjugated antibodies. Examples include At 211 , 1 131 , 1 125 , Y 90 , Re 186 , Re 188 ,
- the conjugate when used for detection, it may comprise a radioactive atom for scintigraphic studies, for example tc99m or 1123, or a spin label for nuclear magnetic resonance (NMR) imaging (also known as magnetic resonance imaging, mri), such as iodine- 123 again, iodine-131, indium-I l l, fluorine- 19, carbon-13, nitrogen- 15, oxygen- 17, gadolinium, manganese or iron.
- NMR nuclear magnetic resonance
- the radio- or other labels may be incorporated in the conjugate in known ways.
- the peptide may be biosynthesized or may be synthesized by chemical amino acid synthesis using suitable amino acid precursors involving, for example, fluorine- 19 in place of hydrogen.
- Labels such as tc 99 m or l 123 , Re 186 , Re 188 and In 111 can be attached via a cysteine residue in the peptide.
- Yttrium-90 can be attached via a lysine residue.
- the IODOGEN method (Fraker et al (1978) Biochem. Biophys. Res. Commun. 80: 49-57) can be used to incorporate iodine-123.
- Conjugates of the antibody and cytotoxic agent may be made using a variety of bifunctional protein coupling agents such as N-succinimidyl-3-(2-pyridyldithio) propionate (SPDP), succinimidyl-4-(N-maleimidomethyl) cyclohexane-l-carboxylate (SMCC), iminothiolane (IT), bifunctional derivatives of imidoesters (such as dimethyl adipimidate HC1), active esters (such as disuccinimidyl suberate), aldehydes (such as glutaraldehyde), bis-azido compounds (such as bis (p-azidobenzoyl) hexanediamine), bis-diazonium derivatives (such as bis-(p-diazoniumbenzoyl)-ethylenediamine), diisocyanates (such as toluene 2,6-diisocyanate), and bis-active fluorine compounds (such
- a ricin immunotoxin can be prepared as described in Vitetta et al., Science 238: 1098 (1987).
- Carbon- 14-labeled l-isothiocyanatobenzyl-3-methyldiethylene triaminepentaacetic acid (MX-DTPA) is an exemplary chelating agent for conjugation of radionucleotide to the antibody. See W094/11026.
- the linker may be a "cleavable linker" facilitating release of the cytotoxic drug in the cell.
- an acid-labile linker for example, an acid-labile linker, peptidase-sensitive linker, photolabile linker, dimethyl linker or disulfide-containing linker (Chari et al., Cancer Research 52: 127-131 (1992); U.S. Patent No. 5,208,020) may be used.
- the compounds expressly contemplate, but are not limited to, ADC prepared with cross-linker reagents: BMPS, EMCS, GMBS, HBVS, LC-SMCC, MBS, MPBH, SBAP, SIA, SIAB, SMCC, SMPB, SMPH, sulfo-EMCS, sulfo-GMBS, sulfo-KMUS, sulfo-MBS, sulfo-SIAB, sulfo-SMCC, and sulfo-SMPB, and SVSB (succinimidyl-(4- vinylsulfone)benzoate) which are commercially available (e.g., from Pierce
- an antibody is conjugated to one or more drug moieties (D), e.g. about 1 to about 20 drug moieties per antibody, through a linker (L).
- the ADC of Formula I may be prepared by several routes, employing organic chemistry reactions, conditions, and reagents known to those skilled in the art, including: (1) reaction of a nucleophilic group of an antibody with a bivalent linker reagent, to form Ab-L, via a covalent bond, followed by reaction with a drug moiety D; and (2) reaction of a nucleophilic group of a drug moiety with a bivalent linker reagent, to form D-L, via a covalent bond, followed by reaction with the nucleophilic group of an antibody. Additional methods for preparing ADC are described herein.
- the linker may be composed of one or more linker components.
- exemplary linker components include 6-maleimidocaproyl ("MC”), maleimidopropanoyl ("MP”), valine-citrulline (“val-cit”), alanine-phenylalanine (“ala-phe”), p- aminobenzyloxycarbonyl (“PAB”), N-Succinimidyl 4-(2-pyridylthio) pentanoate (“SPP”), N-Succinimidyl 4-(N-maleimidomethyl) cyclohexane-1 carboxylate
- SMCC SMCC
- SIAB N-Succinimidyl (4-iodo-acetyl) aminobenzoate
- Additional linker components are known in the art and some are described herein. See also “Monomethylvaline Compounds Capable of Conjugation to Ligands", US Ser. No. 10/983,340, filed Nov. 5, 2004, the contents of which are hereby incorporated by reference in its entirety.
- the linker may comprise amino acid residues.
- Exemplary amino acid linker components include a dipeptide, a tripeptide, a tetrapeptide or a pentapeptide.
- Exemplary dipeptides include: valine-citrulline (vc or val-cit), alanine -phenylalanine (af or ala-phe).
- Exemplary tripeptides include:
- glycine-valine-citrulline gly-val-cit
- glycine-glycine-glycine gly-glycine-glycine
- Amino acid residues which comprise an amino acid linker component include those occurring naturally, as well as minor amino acids and non-naturally occurring amino acid analogs, such as citrulline.
- Amino acid linker components can be designed and optimized in their selectivity for enzymatic cleavage by a particular enzymes, for example, a tumor-associated protease, cathepsin B, C and D, or a plasmin protease.
- Nucleophilic groups on antibodies include, but are not limited to: (i) N- terminal amine groups, (ii) side chain amine groups, e.g. lysine, (iii) side chain thiol groups, e.g. cysteine, and (iv) sugar hydroxyl or amino groups where the antibody is glycosylated.
- Amine, thiol, and hydroxyl groups are nucleophilic and capable of reacting to form covalent bonds with electrophilic groups on linker moieties and linker reagents including: (i) active esters such as NHS esters, HOBt esters, haloformates, and acid halides; (ii) alkyl and benzyl halides such as haloacetamides; (iii) aldehydes, ketones, carboxyl, and maleimide groups. Certain antibodies have reducible interchain disulfides, i.e. cysteine bridges. Antibodies may be made reactive for conjugation with linker reagents by treatment with a reducing agent such as DTT (dithiothreitol).
- a reducing agent such as DTT (dithiothreitol).
- Each cysteine bridge will thus form, theoretically, two reactive thiol nucleophiles.
- Additional nucleophilic groups can be introduced into antibodies through the reaction of lysines with 2-iminothiolane (Traut's reagent) resulting in conversion of an amine into a thiol.
- Reactive thiol groups may be introduced into the antibody (or fragment thereof) by introducing one, two, three, four, or more cysteine residues ⁇ e.g., preparing mutant antibodies comprising one or more non-native cysteine amino acid residues).
- Antibody drug conjugates may also be produced by modification of the antibody to introduce electrophilic moieties, which can react with nucleophilic substituents on the linker reagent or drug.
- the sugars of glycosylated antibodies may be oxidized, e.g. with periodate oxidizing reagents, to form aldehyde or ketone groups which may react with the amine group of linker reagents or drug moieties.
- the resulting imine Schiff base groups may form a stable linkage, or may be reduced, e.g. by borohydride reagents to form stable amine linkages.
- reaction of the carbohydrate portion of a glycosylated antibody with either glactose oxidase or sodium meta-periodate may yield carbonyl (aldehyde and ketone) groups in the protein that can react with appropriate groups on the drug (Hermanson, Bioconjugate Techniques).
- proteins containing N-terminal serine or threonine residues can react with sodium meta-periodate, resulting in production of an aldehyde in place of the first amino acid (Geoghegan & Stroh, (1992) Bioconjugate Chem. 3: 138-146; US 5362852).
- Such aldehyde can be reacted with a drug moiety or linker nucleophile.
- nucleophilic groups on a drug moiety include, but are not limited to: amine, thiol, hydroxyl, hydrazide, oxime, hydrazine, thiosemicarbazone, hydrazine carboxylate, and arylhydrazide groups capable of reacting to form covalent bonds with electrophilic groups on linker moieties and linker reagents including: (i) active esters such as NHS esters, HOBt esters, haloformates, and acid halides; (ii) alkyl and benzyl halides such as haloacetamides; (iii) aldehydes, ketones, carboxyl, and maleimide groups.
- a fusion protein comprising the antibody and cytotoxic agent may be made, e.g., by recombinant techniques or peptide synthesis.
- the length of DNA may comprise respective regions encoding the two portions of the conjugate either adjacent one another or separated by a region encoding a linker peptide which does not destroy the desired properties of the conjugate.
- the antibody may be conjugated to a "receptor" (such streptavidin) for utilization in tumor pre -targeting wherein the antibody-receptor conjugate is administered to the patient, followed by removal of unbound conjugate from the circulation using a clearing agent and then administration of a "ligand” ⁇ e.g. , avidin) which is conjugated to a cytotoxic agent ⁇ e.g., a radionucleotide).
- a "receptor” such streptavidin
- antibodies of the invention are useful for detecting the presence of mutant SMO in a biological sample.
- detecting encompasses quantitative or qualitative detection.
- a biological sample comprises a cell or tissue, such as tumor tissue.
- the invention provides a method of detecting the presence of mutant SMO in a biological sample.
- the method comprises contacting the biological sample with an anti-mutant SMO antibody under conditions permissive for binding of the anti-mutant SMO antibody to mutant SMO, and detecting whether a complex is formed between the anti-mutant SMO antibody and mutant SMO.
- the invention provides a method of diagnosing a disorder associated with expression of mutant SMO.
- the method comprises contacting a test cell with an anti-mutant SMO antibody; determining the level of expression (either quantitatively or qualitatively) of mutant SMO by the test cell by detecting binding of the anti-mutant SMO antibody to mutant SMO; and comparing the level of expression of mutant SMO by the test cell with the level of expression of mutant SMO by a control cell (e.g., a normal cell of the same tissue origin as the test cell or a cell that expresses wild-type SMO at levels comparable to such a normal cell), wherein a higher level of expression of mutant SMO by the test cell as compared to the control cell indicates the presence of a disorder associated with increased expression of mutant SMO.
- the test cell is obtained from an individual suspected of having a disorder associated with increased expression of mutant SMO.
- the disorder is a cell proliferative disorder, such as a cancer or a tumor.
- Exemplary disorders that may be diagnosed using an antibody of the invention include, but are not limited to medulloblastoma, pancreatic cancer basal cell carcinoma.
- Certain other methods can be used to detect binding of antibodies to mutant SMO. Such methods include, but are not limited to, antigen-binding assays that are well known in the art, such as western blots, radioimmunoassays, ELISA (enzyme linked immunosorbent assay), "sandwich” immunoassays, immunoprecipitation assays, fluorescent immunoassays, protein A immunoassays, and
- labels include, but are not limited to, labels or moieties that are detected directly (such as fluorescent, chromophoric, electron-dense, chemiluminescent, and radioactive labels), as well as moieties, such as enzymes or ligands, that are detected indirectly, e.g., through an enzymatic reaction or molecular interaction.
- exemplary labels include, but are not
- radioisotopes P, C, I, H, and I fluorophores such as rare earth chelates or fluorescein and its derivatives, rhodamine and its derivatives, dansyl, umbelliferone, luceriferases, e.g., firefly luciferase and bacterial luciferase (U.S. Pat. No.
- luciferin 2,3-dihydrophthalazinediones
- horseradish peroxidase HRP
- alkaline phosphatase alkaline phosphatase
- ⁇ -galactosidase glucoamylase
- lysozyme saccharide oxidases, e.g., glucose oxidase, galactose oxidase, and glucose-6-phosphate dehydrogenase
- heterocyclic oxidases such as uricase and xanthine oxidase, coupled with an enzyme that employs hydrogen peroxide to oxidize a dye precursor such as HRP, lactoperoxidase, or microperoxidase, biotin/avidin, spin labels, bacteriophage labels, stable free radicals, and the like.
- antibodies are immobilized on an insoluble matrix. Immobilization may entail separating an anti-mutant SMO antibody from any mutant SMO that remains free in solution. This conventionally is accomplished by either insolubilizing the anti-mutant SMO antibody before the assay procedure, as by adsorption to a water-insoluble matrix or surface (Bennich et al., U.S. 3,720,760), or by covalent coupling (for example, using glutaraldehyde cross-linking), or by insolubilizing the anti-mutant SMO antibody after formation of a complex between the anti-mutant SMO antibody and mutant SMO, e.g., by immunoprecipitation.
- nucleic acid probesas described herein are useful for detecting the presence of mutant SMO nucleic acid in a biological sample.
- detecting encompasses quantitative or qualitative detection.
- a biological sample comprises a cell or tissue, such as tumor tissue.
- the invention provides a method of detecting the presence of mutant SMO-necoding nucleic acid in a biological sample.
- the method comprises contacting nucleic acid from the biological sample with a probe as described herein and hybridizing the probe to the nucleic acid under conditions permissive for hybridization under stringent conditions, and detecting whether a complex is formed between the probe and the nucleic acid sample.
- the mutant SMO-encoding nucleic acid may be detected using any combination thereof
- detection of a mutant SMO as described herein cell indicates the presence of a disorder associated with increased expression of mutant SMO (i.e., resistance to treatment with a Smo inhibitor such as GDC-0449).
- the test cell is obtained from an individual suspected of having a resistant tumor associated with expression of mutant SMO.
- Exemplary disorders that may be diagnosed using an antibody of the invention include, but are not limited to medulloblastoma, pancreatic cancer basal cell carcinoma.
- Mutant SMO may be detected in cell based assays as known in the art including, but not limited to binding of a mutant SMO-detecting antibody to the surface of a cell sample, such as a tumor sample in vitro Immunohistochemical staining of histological preparations of tumor samples or tissue suspected of containing mutant SMO.
- D Methods of screening for compounds that bind to mutant SMO
- the invention provides a method for screening for compounds that bind to mutant SMO. Without being held to any particular mode of operation, it is expected that much in the way that GDC-0449 binds wild-type SMO and doesn't bind mutant SMO, a compound which acts as an inhibitor of mutant SMO would dind mutant SMO in the same region within the carboxy-terminal portion of the transmembrane domain NO. 6 (TM6). Thus, one may express this region of the mutant SMO proteinand run binding assays using a library of compounds by any means known in the art. Also one may use a smaller library of compounds represented by variations of GDC-0449 using a modeling approach basedon potential contact points of GDC-0449 and then modeling similar contact points for mutant SMO and variations of GDC- 0449.
- Such modeling programs and algorithms may be any that are known in the art.
- Compounds that bind mutant SMO and wild-type SMO may be identified that are inhibitors of both wild-type and mutant SMO.
- compounds may be discovered that bind to mutant SMO, but which do not bind to wild-type SMo and therefore are inhibitors only for mutant SMO.
- the compounds to be screened are small molecule compounds such as variants of GDC-0449.
- the compounds that bind mutant SMO are antibodies that specifically recognize an epitope that is in the same region as the binding site of GDC-0449 to wild-type SMO.
- the antibody binds to a region in the carboxy-terminal portion of TM6 of mutant SMO and inhibits mutant SMO activity.
- Compounds may alternatively, or additionally be screened for their ability to inhibit mutant SMO activity.
- These assays may be performed in cells that have a hedgehog signaling pathway intact but which express a recombinant SMO bearing the mutation in place of, or in addition to wild-type SMO.
- the cells express both wild-type and mutant SMO and are incubated with GDC-0449 and a candidiate inhibitor.
- the cells express only mutant SMo and may be incubated with Hh and the candidate inhibitor alone (i.e., in the absence of GDC-0449).
- the compound is an inhibitor of mutant SMO if Hh signaling is reduced or inhibited in such cells.
- the invention provides methods of treating a patient in having a hedgehog signaling-dependent tumor that is resistant to chemotherapeutic compounds such as GDC-0449 with a compound that binds a mutant SMO.
- an antibody of the invention may be used in, for example, in vitro, ex vivo, and in vivo therapeutic methods.
- the invention provides methods for treating cancer, inhibiting unwanted cellular proliferation, inhibiting metastasis of cancer and inducing apoptosis of tumor cells either in vivo or in vitro, the method comprising exposing a cell to an antibody of the invention under conditions permissive for binding of the antibody to mutant SMO.
- the cell is a myelogenous leukemia cell, a lung cancer cell, a gastric cancer cell, a breast cancer cell, a prostate cancer cell, a renal cell cancer cell, and a glioblastoma cell.
- an antibody of the invention can be used for inhibiting an activity of mutant SMO, the method comprising exposing mutant SMO to an antibody of the invention such that the activity of mutant SMO is inhibited.
- the invention provides methods for treating cancer comprising administering to an individual an effective amount of an antibody of the invention.
- a method for treating cancer comprises administering to an individual an effective amount of a pharmaceutical formulation comprising an antibody of the invention and, optionally, at least one additional therapeutic agent, such as those provided below.
- Antibodies of the invention can be used either alone or in combination with other compositions in a therapy.
- an antibody of the invention may be co-administered with at least one additional therapeutic agent and/or adjuvant.
- an additional therapeutic agent is an anti-VEGF antibody.
- Such combination therapies noted above encompass combined administration (where two or more therapeutic agents are included in the same or separate formulations), and separate administration, in which case, administration of the antibody of the invention can occur prior to, simultaneously, and/or following, administration of the additional therapeutic agent and/or adjuvant.
- Antibodies of the invention can also be used in combination with radiation therapy.
- an antibody of the invention is used in a method for binding mutant SMO in an individual suffering from a disorder associated with increased mutant SMO expression and/or activity, the method comprising
- mutant SMO in the individual is bound.
- the mutant SMO is human mutant SMO, and the individual is human.
- An antibody of the invention (and any additional therapeutic agent or adjuvant) can be administered by any suitable means, including parenteral, subcutaneous, intraperitoneal, intrapulmonary, and intranasal, and, if desired for local treatment, intralesional administration.
- Parenteral infusions include intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous administration.
- the antibody is suitably administered by pulse infusion, particularly with declining doses of the antibody. Dosing can be by any suitable route, e.g. by injections, such as intravenous or subcutaneous injections, depending in part on whether the
- an antibody of the invention can be expressed intracellularly as an intrabody.
- intrabody refers to an antibody or antigen-binding portion thereof that is expressed intracellularly and that is capable of selectively binding to a target molecule, as described, e.g., in Marasco, Gene Therapy 4: 11-15 (1997); Kontermann, Methods 34: 163-170 (2004); U.S. Patent Nos.
- Intracellular expression of an intrabody may be effected by introducing a nucleic acid encoding the desired antibody or antigen-binding portion thereof (lacking the wild-type leader sequence and secretory signals normally associated with the gene encoding that antibody or antigen-binding fragment) into a target cell.
- a nucleic acid encoding the desired antibody or antigen-binding portion thereof lacking the wild-type leader sequence and secretory signals normally associated with the gene encoding that antibody or antigen-binding fragment
- One or more nucleic acids encoding all or a portion of an antibody of the invention can be delivered to a target cell, such that one or more intrabodies are expressed which are capable of binding to an intracellular target polypeptide and modulating the activity of the target polypeptide.
- Any standard method of introducing nucleic acids into a cell may be used, including, but not limited to, microinjection, ballistic injection, electroporation, calcium phosphate precipitation, liposomes, and transfection with retroviral, adenoviral, adeno-associated viral and vaccinia vectors carrying the nucleic acid of interest.
- nucleic acid may be introduced into a patient's cells by in vivo and ex vivo methods.
- nucleic acid is injected directly into the patient, e.g., at the site where therapeutic intervention is required.
- nucleic acid is introduced into a cell using transfection with viral vectors (such as adenovirus, Herpes simplex I virus, or adeno-associated virus) and lipid-based systems (useful lipids for lipid-mediated transfer of the gene are DOTMA, DOPE and DC-Choi, for example).
- viral vectors such as adenovirus, Herpes simplex I virus, or adeno-associated virus
- lipid-based systems useful lipids for lipid-mediated transfer of the gene are DOTMA, DOPE and DC-Choi, for example.
- nucleic acid is introduced into those isolated cells, and the modified cells are administered to the patient either directly or, for example, encapsulated within porous membranes which are implanted into the patient (see, e.g., U.S. Patent Nos. 4,892,538 and 5,283,187).
- a commonly used vector for ex vivo delivery of a nucleic acid is a retroviral vector.
- Such peptides can be synthesized chemically and/or produced by recombinant DNA technology. See, e.g., Marasco et ah, Proc. Natl. Acad. Sci. USA, 90: 7889-7893 (1993).
- certain embodiments of the invention provide for the antibody to traverse the blood-brain barrier.
- Several art-known approaches exist for transporting molecules across the blood-brain barrier including, but not limited to, physical methods, lipid-based methods, stem cell-based methods, and receptor and channel-based methods.
- Circumvention methods include, but are not limited to, direct injection into the brain (see, e.g., Papanastassiou et ah, Gene Therapy 9: 398-406 (2002)), interstitial infusion/convection-enhanced delivery (see, e.g., Bobo et al, Proc. Natl. Acad. Sci. USA 91 : 2076-2080 (1994)), and implanting a delivery device in the brain (see, e.g., Gill et al., Nature Med.
- Methods of creating openings in the barrier include, but are not limited to, ultrasound (see, e.g., U.S. Patent Publication No. 2002/0038086), osmotic pressure ⁇ e.g., by administration of hypertonic mannitol (Neuwelt, E. A., Implication of the Blood-Brain Barrier and its Manipulation, Vols 1 & 2, Plenum Press, N.Y. (1989)), permeabilization by, e.g., bradykinin or
- permeabilizer A-7 see, e.g., U.S. Patent Nos. 5,112,596, 5,268,164, 5,506,206, and 5,686,416), and transfection of neurons that straddle the blood-brain barrier with vectors containing genes encoding the antibody (see, e.g., U.S. Patent Publication No. 2003/0083299).
- Lipid-based methods of transporting an antibody across the blood-brain barrier include, but are not limited to, encapsulating the antibody in liposomes that are coupled to antibody binding fragments that bind to receptors on the vascular endothelium of the blood-brain barrier (see, e.g., U.S. Patent Application Publication No. 20020025313), and coating the antibody in low-density lipoprotein particles (see, e.g., U.S. Patent Application Publication No. 20040204354) or apolipoprotein E (see, e.g., U.S. Patent Application Publication No. 20040131692).
- NPCs neural progenitor cells
- Receptor and channel-based methods of transporting an antibody across the blood-brain barrier include, but are not limited to, using glucocorticoid blockers to increase permeability of the blood-brain barrier (see, e.g., U.S. Patent Application Publication Nos. 2002/0065259, 2003/0162695, and 2005/0124533); activating potassium channels (see, e.g., U.S. Patent Application Publication No.
- Factors for consideration in this context include the particular disorder being treated, the particular mammal being treated, the clinical condition of the individual patient, the cause of the disorder, the site of delivery of the agent, the method of administration, the scheduling of administration, and other factors known to medical practitioners.
- the antibody need not be, but is optionally formulated with one or more agents currently used to prevent or treat the disorder in question.
- the effective amount of such other agents depends on the amount of antibody present in the formulation, the type of disorder or treatment, and other factors discussed above. These are generally used in the same dosages and with administration routes as described herein, or about from 1 to 99% of the dosages described herein, or in any dosage and by any route that is
- an antibody of the invention when used alone or in combination with one or more other additional therapeutic agents, will depend on the type of disease to be treated, the type of antibody, the severity and course of the disease, whether the antibody is
- the antibody is suitably administered to the patient at one time or over a series of treatments.
- about 1 ⁇ g/kg to 15 mg/kg (e.g. O.lmg/kg-lOmg/kg) of antibody can be an initial candidate dosage for administration to the patient, whether, for example, by one or more separate administrations, or by continuous infusion.
- One typical daily dosage might range from about 1 ⁇ g/kg to 100 mg/kg or more, depending on the factors mentioned above.
- the treatment would generally be sustained until a desired suppression of disease symptoms occurs.
- One exemplary dosage of the antibody would be in the range from about 0.05 mg/kg to about 10 mg/kg.
- one or more doses of about 0.5 mg/kg, 2.0 mg/kg, 4.0 mg/kg or 10 mg/kg (or any combination thereof) may be administered to the patient.
- Such doses may be administered intermittently, e.g. every week or every three weeks (e.g. such that the patient receives from about two to about twenty, or e.g. about six doses of the antibody).
- An initial higher loading dose, followed by one or more lower doses may be administered.
- An exemplary dosing regimen comprises administering an initial loading dose of about 4 mg/kg, followed by a weekly maintenance dose of about 2 mg/kg of the antibody.
- other dosage regimens may be useful. The progress of this therapy is easily monitored by conventional techniques and assays.
- small molecule compounds that may be used to treat GDC-0449- resistant tumors due to a mutation in smoothened at amino acid position 473 are the following:
- the small molecule is provided in an effective amount to inhibit mutant SMO activity without causing untoward effects on the subject to whom the compound is administered.
- the compound may be administered by any suitable means, including parenteral, subcutaneous, intraperitoneal, intrapulmonary, and intranasal, and, if desired for local treatment, intralesional administration.
- Parenteral infusions include intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous
- Dosing can be by any suitable route, e.g. by injections, such as intravenous or subcutaneous injections, depending in part on whether the
- PI3K inhibitors include any known in the art, including but not limited to those described in Maira S-M et al. (2009) "PI3K Inhibitors for Cancer Treament: Where Do We Stand?" Biochem. Soc. Trans. 37:265-272.
- GDC- 0449 targets the G protein coupled- like receptor, Smoothened (SMO), which becomes activated following loss of PTCHl (Charles M. Rudin et al. (2009) N. Engl. J. Med. 361 : 1173-1178; Molckovsky, A. and L.L. Siu (2008) J. Hematol. Oncol. 1 :20).
- SMO G protein coupled- like receptor
- the PTCHl -W844C mutation was not capable of suppressing SMO activity in a Hh-responsive, GLI-luciferase reporter cell line (C3H10T1 ⁇ 2 fibroblasts) when co-transfected together with wild-type (WT) SMO, indicating that this specific mutation can inhibit the ability of PTCHl to repress SMO and thus lead to aberrant, ligand-independent activation of the Hh pathway (Fig. 5).
- PET scans taken ⁇ 3 months following initiation of treatment indicated disease progression.
- KAAD-Cyclopamine was purchased from Toronto
- Hedgehog Pathway gene status Exons covering the open reading frame of mouse and human SMO/SMO, in addition to exon 15 of PTCHI, were PCR-amplified from genomic DNA using a pair of nested primers.
- the internal pair of primers used in the amplification contained ml 3 forward or ml 3 reverse primer sequences.
- free nucleotides and excess primer were removed using ExoSAP-IT kit (USB); PCR products were sequenced in both directions using ml 3 sequencing primers. PCR products were cycle-sequenced using BigTerminator Kit (Applied Biosystems). All sequencing products were resolved on a 3730x1 sequencing machine (Applied Biosystems).
- Sequence trace files were analyzed using Sequencher (GeneCodes) and/or Mutation Surveyor (SoftGenetics LLC).
- the SMO D473H mutation was additionally confirmed by primer extension and MALDI-TOF mass spectroscopy of the amplified DNA (MassARRAY, Sequenom, San Diego, California).
- the following primers were utilized: Extend Primer (UEP.D473H): TCAGCTGCCACTTCTAC (5081.3 Da) (SEQ ID NO: 13); Analyte G: TCAGCTGCCACTTCTACG (5368.5 Da) (SEQ ID NO: 14); Analyte C: TCAGCTGCCACTTCTACC (5328.5 Da) (SEQ ID NO: 15).
- the SMO D473H mutation was previously reported as a rare SNP (ref SNP ID: rs 17710891), however we were unable to confirm this genotype in DNA obtained from the affected individual or their pedigree.
- the region surrounding the SMO exon 8 mutational site was PCR amplified from the primary disease biopsy DNA, metastatic disease biopsy DNA and a control normal DNA (Promega, WI), pooled and analyzed on an Illumina Genome Analyzer.
- a sequence barcode 'AACGCA' for the primary disease DNA, 'AACTGC for metastatic disease DNA and 'AAGCCT' for normal DNA was added as part of the PCR process and this sequence was used to sort the sequences into the three categories post sequencing.
- a total of 57 million 36-bp reads covering the target region was analyzed for the presence of the mutated allele (G>C).
- the background sequencing error rate 0.02%>, which represents the threshold of detection using this technology.
- a binomial test (p ⁇ 0) excludes the presence of the mutant allele at a 0.1% or higher level in any of the samples.
- C3H10T1/2 cells (ATCC, Cat. # CCL-226) were seeded into six -well plates at 1.5 x 10 5 cells/well in DMEM High Glucose with 4 mM glutamine, 10 mM Hepes pH 7.2 and 10% FBS the afternoon before transfection. Cells were transfected the next morning with 400 ng of expression construct, 400 ng of Gli-BS and 200 ng of pRL-TK per well using GeneJuice Transfection Reagent (Novagen, Cat. # 70967). For the PTCH1 inhibition experiments, cells were transfected with an additional 200 ng of DNA containing varying ratios of PTCH1 to eGFP expression constructs.
- NF-KB and SV40 reporter assays Gli-BS was replaced with either pGL4.32 or pGL3-Promoter. Six hours later, cells were collected by trypsin treatment and each well was redistributed over four wells of a 12-well plate. The FBS content of the culture medium was reduced to 0.5% the following morning to induce formation of primary cilia, and small molecule Hh inhibitors were added at indicated concentrations. Luciferase activity was determined 48 hours later with the Dual-Glo Luciferase Assay System (Promega, Cat. # E2940). Values were divided by Renilla luciferase activities to normalize for transfection efficiency and are shown as the mean of three separate experiments ⁇ 1 standard deviation.
- G/z ' -luciferase reporter assays were performed as described above (Rudin, CM. et al. (2009) N. Engl. J. Med. 361 : 1173- 1178) with the following modifications; C3H10T1/2 cells (ATCC, CCL-226) were seeded into six -well plates at 1.85 x 10 5 cells/well and values shown are the mean of four separate experiments ⁇ 1 standard deviation (SD).
- [ 3 H]-GDC-0449-binding assays HEK-293 cells were transfected with SMO expression constructs, harvested, fixed and washed as previously described. Cells were resuspended in PBS, seeded into 96 well plates (2xl0 6 cells/well) and incubated for 1 h at 37°C with 5 nM [ 3 H]-GDC-0449 (0.05 ⁇ ; Tritec, Teufen,
- transfected cells were dislodged in ImM EDTA/PBS and subsequently incubated with SMO antibody, 2D1 1 (lug/ml), followed by 20 min incubations with biotin-SP-conjugated AffiniPure goat anti-mouse IgG (1 : 100, Jackson Immunoresearch Labs 115-065-071) and R-Phycoerythrin-conjugated
- Streptavidin (1 :50, Jackson Immunoresearch Labs 016-110-084). After resuspension in PI (500ng/ml), cells were analyzed using a BD Biosciences HTS FacsCalibur.
- mice on a C57BL/6 background were monitored weekly for the presence of medulloblastomas by MRI. All mice were monitored daily for any signs of abnormal behavior indicative of CNS involvement.
- Mice with well-defined tumors detected by MRI were sacrificed and tumors dissected from normal cerebellum, mechanically dissociated, and 5 xlO 6 cells injected into the lateral thoracic region of CD-I nude mice (CRL). Tumors were allowed to progress to approximately 400 mm in size at which time mice were treated with 75 mg/kg GDC- 0449 (free base equivalents) once daily until tumors decreased in size to
- Tumor-bearing animals were generated via serial subcutaneous propagation of murine Ptch+/-;p53-/- MB tumor lines (Wetmore C. et al. (2001) Cancer Res. 61 :513-516).
- Subcutaneous tumors 1500-2000 mm3 were excised from donor mice under aseptic conditions, minced in High Glucose DMEM by repeated slicing and chopping with two #10 scalpels and passed through a cell dissociation sieve (Sigma, CD1-1KT).
- mice were administered orally 0.2 ml of either vehicle twice daily, compound 5 at 100 mg kg-1 once daily, or HhAntag at 100 mg kg-1 twice daily for the HPI study, and either vehicle or drug at 8 to 10 mg kg-1 once daily for the PI3K inhibitor study.
- Mice were euthanized if tumors exceeded 2000 mm and/or if their body weight dropped > 20%>. All mice were housed and maintained according to the animal use guidelines of Genentech Inc., conforming to California State legal and ethical practices.
- RNA isolation and qRT-PCR Total RNA was extracted from tumors using the RNeasy Mini Kit (Qiagen 74106). RNA concentration was determined with a NanoDrop spectrophotometer and qRT-PCR was carried out with 100 ng RNA on an Applied Biosystems 7500 theromocycler. Expression levels were normalized to Rpll9 and are presented as normalized gene expression values (2-ACt).
- a TaqMan gene expression assay for GUI was purchased from Applied Biosystems, for which the probe (Assay ID: Mm00494646_gl) spanned the exon 3-4 boundary.
- the primer and probe sequences fox Rpll9 are F: 5 '-AGAAGGTGACCTGGATGAGA-3 ' (SEQ ID NO: 10), R: 5'-TGATACATATGGCGGTC AATCT-3' (SEQ ID NO: 11) and P: 5'- CTTCTCAGGAGATACCGGGAATC CAAG-3' (SEQ ID NO: 12).
- Smo immunostaining S12 cells were plated to confluency and serum-starved for 16 h ⁇ 200 ng/ml octyl-Shh in the presence of saturating compound levels (5 ⁇ for cyclopamine, ⁇ for the others). Cells were then fixed in 100% methanol, stained with anti-Smo (5928B, a rabbit pAb raised against the C-terminal tail of mouse Smo (Wen X. et al. (2010) Mol. Cell. Biol.
- Genomic DNA was isolated from tumors with the AllPrep DNA/RNA Mini Kit (Qiagen) and every exon from murine Smo, Sufu and GU2 was PCR-amp lifted using a pair of nested primers containing Ml 3 forward and reverse sequences. Excess primers and free nucleotides were removed with the ExoSAP-IT kit (USB) and PCR products were cycle-sequenced in both directions using M13 sequencing primers, a BigDye Terminator v3.1 Kit and a 3730x1 DNA analyzer (both Applied Biosystems). Sequence files were analyzed using Sequencher (GeneCodes) and Mutation Surveyor (SoftGenetics LLC) software.
- GU2 F 5'- GC AGGAC ATTCC ACAC AGTTCTTG-3 ' (SEQ ID NO:4)
- GU2 R 5 '-ATAGGTGCTGGGATACAGGCTTG-3 ' (SEQ ID NO:5)
- Ccndl F 5'-TACCCTGACACCAATCTCCTCAACG-3' (SEQ ID NO:6), Ccndl R: 5'- GGAATTCCCATCTTCCCAACTCC-3' (SEQ ID NO:7), Sinel F: 5 '-AGATGGCTGAGTGGGTAAAGG-3 ' (SEQ ID NO:8) and Sinel R: 5 ' -GTGGAGGTC AGAGGAC AAACTT-3 ' (SEQ ID NO:9).
- Protein Extraction Reagent containing protease and phosphatase inhibitors. Lysates were separated on 4-12% Bis-Tris gels and proteins were transferred onto PVDF membranes with an iBlot (Invitrogen). Blots were blocked and incubated overnight at 4°C with 5% milk containing one of the following primary antibodies; anti-cyclin Dl (Cell Signaling, #2922), anti-phospho(Ser473)-AKT (Cell Signaling, #4060), anti-total AKT (Cell Signaling #9272), anti-Gli2 (Cho A. et al. (2008) Dev. Biol.
- GDC-0449 inhibited reporter activity at an IC 50 of 20nM in SMO-WT transfected cells, no inhibition was observed in SMO-D473H transfected cells even at concentrations as high as 3 ⁇ ; indicating that this mutation confers resistance to GDC-0449 without affecting its ability to transmit the Hh signal.
- SMO-D473H also impaired the ability of a chemically divergent SMO inhibitor, KAAD-cyclopamine (J. Taipale et al. (2000) Nature 406: 1005), to inhibit GLI-luciferase reporter activity with a 43-fold change in IC50 (Fig. 10).
- GDC-0449 did not suppress Hh signaling in vivo, as
- Topology prediction and structural modeling of SMO map the Asp-473 residue to the C-terminal end of the sixth transmembrane segment (TM6), a position that is highly conserved across SMO orthologs and the related Frizzled family of Wnt receptors (Fig. IB, Fig. 12).
- TM6 sixth transmembrane segment
- the heptahelical structure of SMO is required for binding of cyclopamine (J. Chen K. et al. (2002) Genes Dev. 16:2743) and is the target for ortho- and allosteric GPCR modulators (Goudet et al. (2004) Drug
- D473 is important for SMO activity and inhibition by GDC-0449.
- SMO-D473P Apart from the possibly misfolded SMO-D473P, all mutants induced Hh pathway activity and were less sensitive to GDC-0449 inhibition than SMO-WT.
- the seemingly responsive D473V mutant was partially drug-resistant in a dose response assay (Fig. 18B).
- the SMO-D473E mutant was also resistant to GDC-0449, even though this conservative substitution maintains a negative charge at this position.
- We next confirmed cell surface expression for several of these mutants (Fig. 18C) and tested their ability to bind GDC-0449 (Fig. 13B). Similar to SMO-D473H, resistance to this HPI correlated with a lack of SMO binding.
- D473 could either be directly involved in GDC-0449 binding or could simply be required to maintain the correct SMO conformation for binding.
- the SMO-D473K and SMO-D473R mutants have auto-activating properties and are resistant to inhibition by PTCH1 (Fig. 13C). However, it is unlikely that they will be naturally occurring oncogenic or drug- resistant mutants, since these amino acid substitutions require two nucleotide changes.
- a screen of chemically diverse HPIs identified several SMO-D473H antagonists.
- SMO mutant inhibitors As potential therapeutics for GDC- 0449 resistant tumors, we screened a panel of 53 antagonists (representative compounds are shown in Fig. 14 A) with potent activity against the wild type protein (Fig. 14B). These compounds were either identified in high-throughput screens (both in house and by others) or were generated by hit-to-lead optimization of screening hits using traditional medicinal chemistry methods.
- C3H10T1/2 cells were co-transfected with wild type or mutant SMO expression vectors together with a G/z ' -luciferase reporter construct (Murone M. et al. (1999) Curr Biol.
- Compound 5 inhibits tumor growth mediated by GDC-0449 resistant Smo. It was important to determine whether compound 5 (Formula III) could also inhibit drug resistant Smo in vivo. To this extent, we generated mice with subcutaneous allografts of the murine Ptch +/ ⁇ ;p53 _/ ⁇ MB tumor line SG274, which had been rendered resistant to GDC-0449 due to a D477G amino acid substitution in Smo, the same aspartic residue that was mutated in human SMO. These mice developed
- GDC-0449 has a differential effect on GUI mRNA expression in two additional resistant MB allograft models.
- SG274 was found to carry a mutation in Smo, indicating that additional mechanisms of resistance to GDC-0449 exist in models SG102 and SG152.
- mutations in the tumor suppressor SUFU predispose individuals to MB (Taylor M.D. et al. (2002) Nat. Genet. 31 :306-310) and could in theory confer resistance to Smo antagonists, neither resistant MB allograft model was mutated in this gene. Naturally, resistance could also occur if these tumors had lost their dependence on Hh signaling.
- Model SGI 02 contained an amplification of a region on chromosome 7 harboring the Hh target gene Ccndl (cyclin Dl), while model SG152 had a high level focal amplification of a region on chromosome 1 encompassing the Hh pathway transcription factor Gli2 (Fig. 20). Both models contained additional copy number aberrations that previously have not been associated with either MB or abnormal Hh signaling (data not shown).
- cyclin Dl and Gli2 act downstream of Smo and have previously been implicated in the development of MB, we formally cannot rule out involvement of these other genomic alterations in GDC-0449 resistance.
- tumor lines as expanded tumors exhibited enhanced mRNA expression and elevated protein levels of both cyclin Dl and Gli2 (Fig. 16B and Fig. 16C).
- Cyclin Dl promotes proliferation through its ability to bind to and stimulate both CDK4 and CDK6, leading to phosphorylation of the retinoblastoma protein and entry into the cell cycle (Kim J.K. and J. A. Diehl (2009) J. Cell. Physiol. 220:292- 296).
- Genetic ablation of Ccndl drastically reduces the incidence of MB in Ptch+/- mice (Pogoriler J. et al. (2006) Development 133:3929-3937), whereas enforced expression of cyclin Dl in Ink4c-/-;p53-/- GNPs enabled cells to initiate MBs when injected back into the brains of immunocompromised recipient animals (Zindy F. et al.
- GDC-0449 down regulated cyclin Dl levels in control tumors, consistent with Ccndl being an Hh target gene (Fig. 16D; Zindy F. et al. (2007) Cancer Res. 67:2676-2684). In contrast, cyclin Dl levels remained elevated in SG102 tumors, even though GDC-0449 diminished GUI mRNA levels. High cyclin Dl levels likely sustain tumor cell proliferation in the presence of GDC-0449, as Ccndl expression is no longer reliant on Hh signaling due to the gene amplification.
- GLI2 amplifications have been observed in human MB, they are relatively rare (Northcott P.A. et al. (2009) Nat. Genet. 41 :465-472).
- Gli2 contains an amino-terminal repressor domain that when deleted results in a constitutively active protein with 30 times higher transcriptional activity (Roessler E. et al. (2005) Hum. Mol. Genet. 14:2181-2188). Tissue specific expression of this truncated transcription factor can lead to MB when ciliogenesis is impaired (Han Y.G. et al. (2009) Nat Med. 15: 1062-1065).
- HPI resistant MB allografts are sensitive to PI3K inhibition. Given the identification of resistance mechanisms downstream of SMO, we looked at other signaling pathways implicated in MB to see if targeting any of these might be an alternative therapeutic approach to combating GDC-0449 resistance. Abnormal phosphoinositide3 -kinase (PI3K)/AKT signaling promotes tumor growth and survival of many human cancers, including MB (Vivanco I., and C.L. Sawyers (2002) Nat.
- PI3K Abnormal phosphoinositide3 -kinase
- PI3K pathway modulation was accompanied by GDC-0941 treatment decreased pAKT and pS6 levels (Fig. 17A). Consequently, pharmacologic inhibition of PI3K/AKT signaling represents a promising therapeutic approach to treating HPI-resistant MB.
Abstract
Description
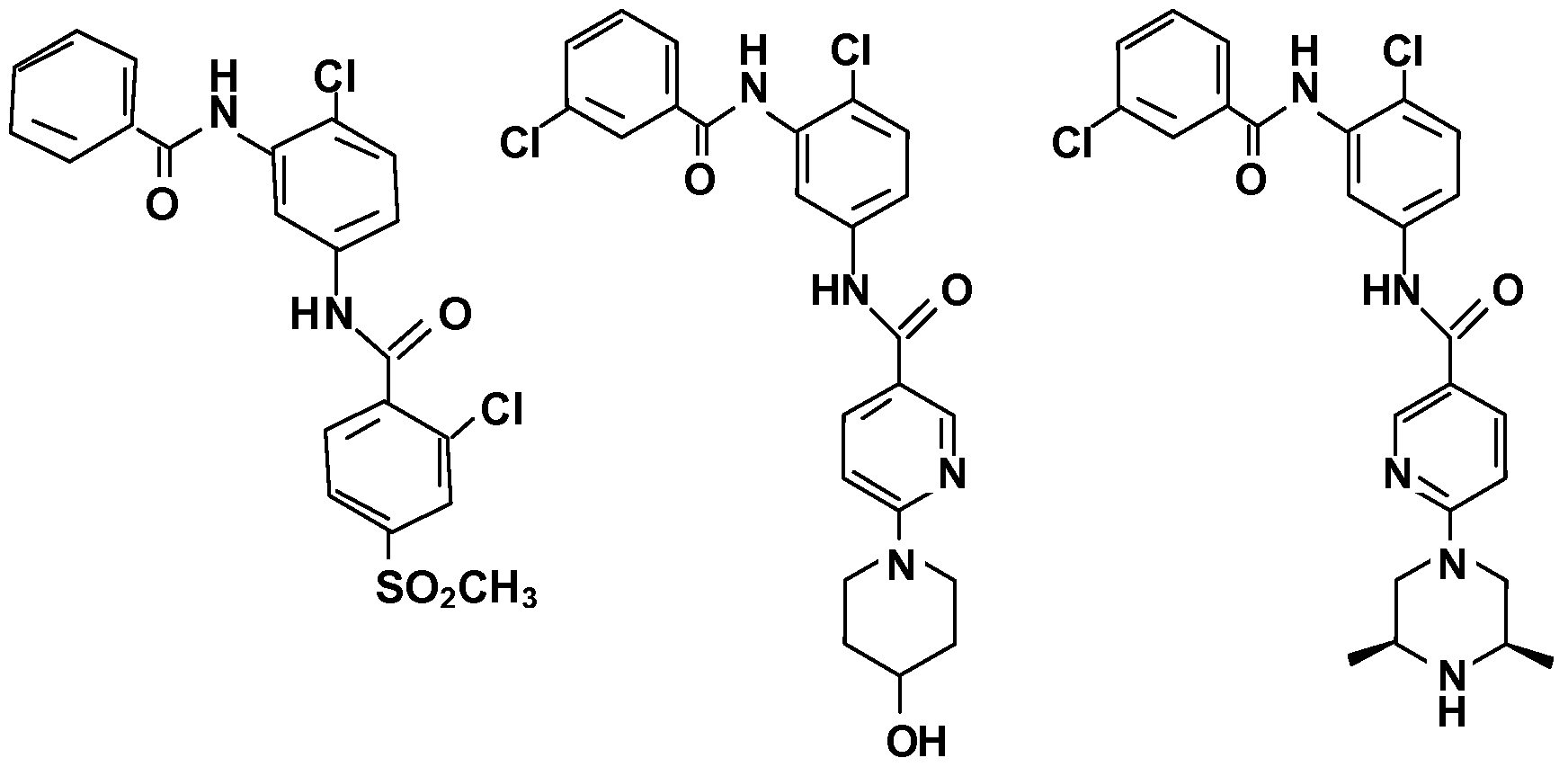


Claims

Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2010289400A AU2010289400B2 (en) | 2009-09-02 | 2010-09-02 | Mutant smoothened and methods of using the same |
| CA2772715A CA2772715C (en) | 2009-09-02 | 2010-09-02 | Mutant smoothened and methods of using the same |
| EP10751758.3A EP2473522B1 (en) | 2009-09-02 | 2010-09-02 | Mutant smoothened and methods of using the same |
| JP2012528067A JP5887270B2 (en) | 2009-09-02 | 2010-09-02 | Mutant SMOOTHENED AND METHOD OF USING THE SAME |
| US13/394,069 US9321823B2 (en) | 2009-09-02 | 2010-09-02 | Mutant smoothened and methods of using the same |
| DK10751758.3T DK2473522T3 (en) | 2009-09-02 | 2010-09-02 | Smoothened MUTANT AND METHODS OF USING THE SAME |
| ES10751758.3T ES2599076T3 (en) | 2009-09-02 | 2010-09-02 | Smoothened mutant and methods of use thereof |
| US15/130,223 US9910050B2 (en) | 2009-09-02 | 2016-04-15 | Mutant smoothened and methods of using the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US23936409P | 2009-09-02 | 2009-09-02 | |
| US61/239,364 | 2009-09-02 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/394,069 A-371-Of-International US9321823B2 (en) | 2009-09-02 | 2010-09-02 | Mutant smoothened and methods of using the same |
| US15/130,223 Division US9910050B2 (en) | 2009-09-02 | 2016-04-15 | Mutant smoothened and methods of using the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011028950A1 true WO2011028950A1 (en) | 2011-03-10 |
Family
ID=42938479
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/047739 WO2011028950A1 (en) | 2009-09-02 | 2010-09-02 | Mutant smoothened and methods of using the same |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US9321823B2 (en) |
| EP (1) | EP2473522B1 (en) |
| JP (3) | JP5887270B2 (en) |
| AU (1) | AU2010289400B2 (en) |
| CA (1) | CA2772715C (en) |
| DK (1) | DK2473522T3 (en) |
| ES (1) | ES2599076T3 (en) |
| WO (1) | WO2011028950A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8481680B2 (en) | 2010-10-05 | 2013-07-09 | Genentech, Inc. | Mutant smoothened and methods of using the same |
| WO2015116902A1 (en) | 2014-01-31 | 2015-08-06 | Genentech, Inc. | G-protein coupled receptors in hedgehog signaling |
| WO2015120075A2 (en) | 2014-02-04 | 2015-08-13 | Genentech, Inc. | Mutant smoothened and methods of using the same |
| WO2017136558A1 (en) | 2016-02-04 | 2017-08-10 | Curis, Inc. | Mutant smoothened and methods of using the same |
| CN107636170A (en) * | 2015-02-04 | 2018-01-26 | 健泰科生物技术公司 | Saltant type Smoothened and its application method |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2987067A1 (en) | 2015-06-05 | 2016-12-08 | Dana-Farber Cancer Institute, Inc. | Compounds and methods for treating cancer |
Citations (345)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US3720760A (en) | 1968-09-06 | 1973-03-13 | Pharmacia Ab | Method for determining the presence of reagin-immunoglobulins(reagin-ig)directed against certain allergens,in aqueous samples |
| US3896111A (en) | 1973-02-20 | 1975-07-22 | Research Corp | Ansa macrolides |
| US4137230A (en) | 1977-11-14 | 1979-01-30 | Takeda Chemical Industries, Ltd. | Method for the production of maytansinoids |
| US4151042A (en) | 1977-03-31 | 1979-04-24 | Takeda Chemical Industries, Ltd. | Method for producing maytansinol and its derivatives |
| EP0003089A1 (en) | 1978-01-06 | 1979-07-25 | Bernard David | Drier for silkscreen printed sheets |
| US4248870A (en) | 1978-10-27 | 1981-02-03 | Takeda Chemical Industries, Ltd. | Maytansinoids and use |
| US4256746A (en) | 1978-11-14 | 1981-03-17 | Takeda Chemical Industries | Dechloromaytansinoids, their pharmaceutical compositions and method of use |
| US4260608A (en) | 1978-11-14 | 1981-04-07 | Takeda Chemical Industries, Ltd. | Maytansinoids, pharmaceutical compositions thereof and methods of use thereof |
| US4265814A (en) | 1978-03-24 | 1981-05-05 | Takeda Chemical Industries | Matansinol 3-n-hexadecanoate |
| US4294757A (en) | 1979-01-31 | 1981-10-13 | Takeda Chemical Industries, Ltd | 20-O-Acylmaytansinoids |
| US4307016A (en) | 1978-03-24 | 1981-12-22 | Takeda Chemical Industries, Ltd. | Demethyl maytansinoids |
| US4308268A (en) | 1979-06-11 | 1981-12-29 | Takeda Chemical Industries, Ltd. | Maytansinoids, pharmaceutical compositions thereof and method of use thereof |
| US4308269A (en) | 1979-06-11 | 1981-12-29 | Takeda Chemical Industries, Ltd. | Maytansinoids, pharmaceutical compositions thereof and method of use thereof |
| US4309428A (en) | 1979-07-30 | 1982-01-05 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| US4313946A (en) | 1981-01-27 | 1982-02-02 | The United States Of America As Represented By The Secretary Of Agriculture | Chemotherapeutically active maytansinoids from Trewia nudiflora |
| US4315929A (en) | 1981-01-27 | 1982-02-16 | The United States Of America As Represented By The Secretary Of Agriculture | Method of controlling the European corn borer with trewiasine |
| US4317821A (en) | 1979-06-08 | 1982-03-02 | Takeda Chemical Industries, Ltd. | Maytansinoids, their use and pharmaceutical compositions thereof |
| US4322348A (en) | 1979-06-05 | 1982-03-30 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| US4331598A (en) | 1979-09-19 | 1982-05-25 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| USRE30985E (en) | 1978-01-01 | 1982-06-29 | Serum-free cell culture media | |
| US4362663A (en) | 1979-09-21 | 1982-12-07 | Takeda Chemical Industries, Ltd. | Maytansinoid compound |
| US4364866A (en) | 1979-09-21 | 1982-12-21 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| US4371533A (en) | 1980-10-08 | 1983-02-01 | Takeda Chemical Industries, Ltd. | 4,5-Deoxymaytansinoids, their use and pharmaceutical compositions thereof |
| EP0073657A1 (en) | 1981-08-31 | 1983-03-09 | Genentech, Inc. | Preparation of hepatitis B surface antigen in yeast |
| US4419446A (en) | 1980-12-31 | 1983-12-06 | The United States Of America As Represented By The Department Of Health And Human Services | Recombinant DNA process utilizing a papilloma virus DNA as a vector |
| US4424219A (en) | 1981-05-20 | 1984-01-03 | Takeda Chemical Industries, Ltd. | 9-Thiomaytansinoids and their pharmaceutical compositions and use |
| US4426330A (en) | 1981-07-20 | 1984-01-17 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US4450254A (en) | 1980-11-03 | 1984-05-22 | Standard Oil Company | Impact improvement of high nitrile resins |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US4534899A (en) | 1981-07-20 | 1985-08-13 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4587044A (en) | 1983-09-01 | 1986-05-06 | The Johns Hopkins University | Linkage of proteins to nucleic acids |
| EP0183070A2 (en) | 1984-10-30 | 1986-06-04 | Phillips Petroleum Company | Transformation of yeasts of the genus pichia |
| US4601978A (en) | 1982-11-24 | 1986-07-22 | The Regents Of The University Of California | Mammalian metallothionein promoter system |
| US4605735A (en) | 1983-02-14 | 1986-08-12 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| WO1987000195A1 (en) | 1985-06-28 | 1987-01-15 | Celltech Limited | Animal cell culture |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4667025A (en) | 1982-08-09 | 1987-05-19 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| EP0244234A2 (en) | 1986-04-30 | 1987-11-04 | Alko Group Ltd. | Transformation of trichoderma |
| US4737456A (en) | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| US4762779A (en) | 1985-06-13 | 1988-08-09 | Amgen Inc. | Compositions and methods for functionalizing nucleic acids |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| DD266710A3 (en) | 1983-06-06 | 1989-04-12 | Ve Forschungszentrum Biotechnologie | Process for the biotechnical production of alkaline phosphatase |
| US4824941A (en) | 1983-03-10 | 1989-04-25 | Julian Gordon | Specific antibody to the native form of 2'5'-oligonucleotides, the method of preparation and the use as reagents in immunoassays or for binding 2'5'-oligonucleotides in biological systems |
| US4828979A (en) | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| US4835263A (en) | 1983-01-27 | 1989-05-30 | Centre National De La Recherche Scientifique | Novel compounds containing an oligonucleotide sequence bonded to an intercalating agent, a process for their synthesis and their use |
| US4845205A (en) | 1985-01-08 | 1989-07-04 | Institut Pasteur | 2,N6 -disubstituted and 2,N6 -trisubstituted adenosine-3'-phosphoramidites |
| US4876335A (en) | 1986-06-30 | 1989-10-24 | Wakunaga Seiyaku Kabushiki Kaisha | Poly-labelled oligonucleotide derivative |
| US4892538A (en) | 1987-11-17 | 1990-01-09 | Brown University Research Foundation | In vivo delivery of neurotransmitters by implanted, encapsulated cells |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| WO1990003430A1 (en) | 1988-09-23 | 1990-04-05 | Cetus Corporation | Cell culture medium for enhanced cell growth, culture longevity and product expression |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| US4948882A (en) | 1983-02-22 | 1990-08-14 | Syngene, Inc. | Single-stranded labelled oligonucleotides, reactive monomers and methods of synthesis |
| WO1990010048A1 (en) | 1989-02-27 | 1990-09-07 | Kerr-Mcgee Corporation | Solvent extraction process |
| US4958013A (en) | 1989-06-06 | 1990-09-18 | Northwestern University | Cholesteryl modified oligonucleotides |
| WO1990010448A2 (en) | 1989-03-07 | 1990-09-20 | Genentech, Inc. | Covalent conjugates of lipid and oligonucleotide |
| US4965199A (en) | 1984-04-20 | 1990-10-23 | Genentech, Inc. | Preparation of functional human factor VIII in mammalian cells using methotrexate based selection |
| US4970198A (en) | 1985-10-17 | 1990-11-13 | American Cyanamid Company | Antitumor antibiotics (LL-E33288 complex) |
| WO1990013646A1 (en) | 1989-04-28 | 1990-11-15 | Transgene S.A. | Application of novel dna fragments as a coding sequence for a signal peptide for the secretion of mature proteins by recombinant yeast, expression cassettes, transformed yeasts and corresponding process for the preparation of proteins |
| WO1990013641A1 (en) | 1989-05-10 | 1990-11-15 | Sloan-Kettering Institute For Cancer Research | Stably transformed eucaryotic cells comprising a foreign transcribable dna under the control of a pol iii promoter |
| US4975278A (en) | 1988-02-26 | 1990-12-04 | Bristol-Myers Company | Antibody-enzyme conjugates in combination with prodrugs for the delivery of cytotoxic agents to tumor cells |
| EP0402226A1 (en) | 1989-06-06 | 1990-12-12 | Institut National De La Recherche Agronomique | Transformation vectors for yeast yarrowia |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| US4981957A (en) | 1984-07-19 | 1991-01-01 | Centre National De La Recherche Scientifique | Oligonucleotides with modified phosphate and modified carbohydrate moieties at the respective chain termini |
| WO1991000360A1 (en) | 1989-06-29 | 1991-01-10 | Medarex, Inc. | Bispecific reagents for aids therapy |
| US5004697A (en) | 1987-08-17 | 1991-04-02 | Univ. Of Ca | Cationized antibodies for delivery through the blood-brain barrier |
| WO1991004753A1 (en) | 1989-10-02 | 1991-04-18 | Cetus Corporation | Conjugates of antisense oligonucleotides and therapeutic uses thereof |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5013830A (en) | 1986-09-08 | 1991-05-07 | Ajinomoto Co., Inc. | Compounds for the cleavage at a specific position of RNA, oligomers employed for the formation of said compounds, and starting materials for the synthesis of said oligomers |
| WO1991006629A1 (en) | 1989-10-24 | 1991-05-16 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| WO1991010741A1 (en) | 1990-01-12 | 1991-07-25 | Cell Genesys, Inc. | Generation of xenogeneic antibodies |
| US5053394A (en) | 1988-09-21 | 1991-10-01 | American Cyanamid Company | Targeted forms of methyltrithio antitumor agents |
| US5079233A (en) | 1987-01-30 | 1992-01-07 | American Cyanamid Company | N-acyl derivatives of the LL-E33288 antitumor antibiotics, composition and methods for using the same |
| WO1992000373A1 (en) | 1990-06-29 | 1992-01-09 | Biosource Genetics Corporation | Melanin production by transformed microorganisms |
| US5082830A (en) | 1988-02-26 | 1992-01-21 | Enzo Biochem, Inc. | End labeled nucleotide probe |
| US5109124A (en) | 1988-06-01 | 1992-04-28 | Biogen, Inc. | Nucleic acid probe linked to a label having a terminal cysteine |
| US5108921A (en) | 1989-04-03 | 1992-04-28 | Purdue Research Foundation | Method for enhanced transmembrane transport of exogenous molecules |
| US5112963A (en) | 1987-11-12 | 1992-05-12 | Max-Planck-Gesellschaft Zur Foerderung Der Wissenschaften E.V. | Modified oligonucleotides |
| US5112596A (en) | 1990-04-23 | 1992-05-12 | Alkermes, Inc. | Method for increasing blood-brain barrier permeability by administering a bradykinin agonist of blood-brain barrier permeability |
| US5118800A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | Oligonucleotides possessing a primary amino group in the terminal nucleotide |
| US5118802A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | DNA-reporter conjugates linked via the 2' or 5'-primary amino group of the 5'-terminal nucleoside |
| WO1992009690A2 (en) | 1990-12-03 | 1992-06-11 | Genentech, Inc. | Enrichment method for variant proteins with altered binding properties |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5138045A (en) | 1990-07-27 | 1992-08-11 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5149797A (en) | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| WO1993001161A1 (en) | 1991-07-11 | 1993-01-21 | Pfizer Limited | Process for preparing sertraline intermediates |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5214136A (en) | 1990-02-20 | 1993-05-25 | Gilead Sciences, Inc. | Anthraquinone-derivatives oligonucleotides |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5218105A (en) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5220007A (en) | 1990-02-15 | 1993-06-15 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5227170A (en) | 1989-06-22 | 1993-07-13 | Vestar, Inc. | Encapsulation process |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| WO1993016185A2 (en) | 1992-02-06 | 1993-08-19 | Creative Biomolecules, Inc. | Biosynthetic binding protein for cancer marker |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US5254469A (en) | 1989-09-12 | 1993-10-19 | Eastman Kodak Company | Oligonucleotide-enzyme conjugate that can be used as a probe in hybridization assays and polymerase chain reaction procedures |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| WO1993021232A1 (en) | 1992-04-10 | 1993-10-28 | Research Development Foundation | IMMUNOTOXINS DIRECTED AGAINST c-erbB-2 (HER-2/neu) RELATED SURFACE ANTIGENS |
| US5258506A (en) | 1984-10-16 | 1993-11-02 | Chiron Corporation | Photolabile reagents for incorporation into oligonucleotide chains |
| US5262536A (en) | 1988-09-15 | 1993-11-16 | E. I. Du Pont De Nemours And Company | Reagents for the preparation of 5'-tagged oligonucleotides |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5264221A (en) | 1991-05-23 | 1993-11-23 | Mitsubishi Kasei Corporation | Drug-containing protein-bonded liposome |
| US5268164A (en) | 1990-04-23 | 1993-12-07 | Alkermes, Inc. | Increasing blood-brain barrier permeability with permeabilizer peptides |
| US5272250A (en) | 1992-07-10 | 1993-12-21 | Spielvogel Bernard F | Boronated phosphoramidate compounds |
| WO1993025673A1 (en) | 1992-06-04 | 1993-12-23 | The Regents Of The University Of California | In vivo gene therapy with intron-free sequence of interest |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5283187A (en) | 1987-11-17 | 1994-02-01 | Brown University Research Foundation | Cell culture-containing tubular capsule produced by co-extrusion |
| WO1994002935A1 (en) | 1992-07-22 | 1994-02-03 | Sinvent A/S | Method and device for active noise reduction in a local area |
| WO1994004690A1 (en) | 1992-08-17 | 1994-03-03 | Genentech, Inc. | Bispecific immunoadhesins |
| US5292873A (en) | 1989-11-29 | 1994-03-08 | The Research Foundation Of State University Of New York | Nucleic acids labeled with naphthoquinone probe |
| WO1994011026A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Therapeutic application of chimeric and radiolabeled antibodies to human b lymphocyte restricted differentiation antigen for treatment of b cell lymphoma |
| US5317098A (en) | 1986-03-17 | 1994-05-31 | Hiroaki Shizuya | Non-radioisotope tagging of fragments |
| US5319080A (en) | 1991-10-17 | 1994-06-07 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5354844A (en) | 1989-03-16 | 1994-10-11 | Boehringer Ingelheim International Gmbh | Protein-polycation conjugates |
| US5356633A (en) | 1989-10-20 | 1994-10-18 | Liposome Technology, Inc. | Method of treatment of inflamed tissues |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5362852A (en) | 1991-09-27 | 1994-11-08 | Pfizer Inc. | Modified peptide derivatives conjugated at 2-hydroxyethylamine moieties |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| US5371241A (en) | 1991-07-19 | 1994-12-06 | Pharmacia P-L Biochemicals Inc. | Fluorescein labelled phosphoramidites |
| US5391723A (en) | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US5395619A (en) | 1993-03-03 | 1995-03-07 | Liposome Technology, Inc. | Lipid-polymer conjugates and liposomes |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5403711A (en) | 1987-11-30 | 1995-04-04 | University Of Iowa Research Foundation | Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5414077A (en) | 1990-02-20 | 1995-05-09 | Gilead Sciences | Non-nucleoside linkers for convenient attachment of labels to oligonucleotides using standard synthetic methods |
| US5417978A (en) | 1993-07-29 | 1995-05-23 | Board Of Regents, The University Of Texas System | Liposomal antisense methyl phosphonate oligonucleotides and methods for their preparation and use |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| US5446137A (en) | 1993-12-09 | 1995-08-29 | Syntex (U.S.A.) Inc. | Oligonucleotides containing 4'-substituted nucleotides |
| US5451463A (en) | 1989-08-28 | 1995-09-19 | Clontech Laboratories, Inc. | Non-nucleoside 1,3-diol reagents for labeling synthetic oligonucleotides |
| US5455233A (en) | 1989-11-30 | 1995-10-03 | University Of North Carolina | Oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5459127A (en) | 1990-04-19 | 1995-10-17 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5462854A (en) | 1993-04-19 | 1995-10-31 | Beckman Instruments, Inc. | Inverse linkage oligonucleotides for chemical and enzymatic processes |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5466786A (en) | 1989-10-24 | 1995-11-14 | Gilead Sciences | 2'modified nucleoside and nucleotide compounds |
| US5469854A (en) | 1989-12-22 | 1995-11-28 | Imarx Pharmaceutical Corp. | Methods of preparing gas-filled liposomes |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5486603A (en) | 1990-01-08 | 1996-01-23 | Gilead Sciences, Inc. | Oligonucleotide having enhanced binding affinity |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5491133A (en) | 1987-11-30 | 1996-02-13 | University Of Iowa Research Foundation | Methods for blocking the expression of specifically targeted genes |
| WO1996007754A1 (en) | 1994-09-02 | 1996-03-14 | The Scripps Research Institute | Methods for producing antibody libraries using universal or randomized immunoglobulin light chains |
| WO1996007321A1 (en) | 1994-09-06 | 1996-03-14 | The Uab Research Foundation | Methods for modulating protein function in cells using intracellular antibody homologues |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5510475A (en) | 1990-11-08 | 1996-04-23 | Hybridon, Inc. | Oligonucleotide multiple reporter precursors |
| US5512439A (en) | 1988-11-21 | 1996-04-30 | Dynal As | Oligonucleotide-linked magnetic particles and uses thereof |
| US5512667A (en) | 1990-08-28 | 1996-04-30 | Reed; Michael W. | Trifunctional intermediates for preparing 3'-tailed oligonucleotides |
| US5512295A (en) | 1994-11-10 | 1996-04-30 | The Board Of Trustees Of The Leland Stanford Junior University | Synthetic liposomes for enhanced uptake and delivery |
| US5514785A (en) | 1990-05-11 | 1996-05-07 | Becton Dickinson And Company | Solid supports for nucleic acid hybridization assays |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5521291A (en) | 1991-09-30 | 1996-05-28 | Boehringer Ingelheim International, Gmbh | Conjugates for introducing nucleic acid into higher eucaryotic cells |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5525465A (en) | 1987-10-28 | 1996-06-11 | Howard Florey Institute Of Experimental Physiology And Medicine | Oligonucleotide-polyamide conjugates and methods of production and applications of the same |
| US5527528A (en) | 1989-10-20 | 1996-06-18 | Sequus Pharmaceuticals, Inc. | Solid-tumor treatment method |
| US5534259A (en) | 1993-07-08 | 1996-07-09 | Liposome Technology, Inc. | Polymer compound and coated particle composition |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5543152A (en) | 1994-06-20 | 1996-08-06 | Inex Pharmaceuticals Corporation | Sphingosomes for enhanced drug delivery |
| US5543158A (en) | 1993-07-23 | 1996-08-06 | Massachusetts Institute Of Technology | Biodegradable injectable nanoparticles |
| US5545730A (en) | 1984-10-16 | 1996-08-13 | Chiron Corporation | Multifunctional nucleic acid monomer |
| US5545807A (en) | 1988-10-12 | 1996-08-13 | The Babraham Institute | Production of antibodies from transgenic animals |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5547932A (en) | 1991-09-30 | 1996-08-20 | Boehringer Ingelheim International Gmbh | Composition for introducing nucleic acid complexes into higher eucaryotic cells |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US5552540A (en) | 1987-06-24 | 1996-09-03 | Howard Florey Institute Of Experimental Physiology And Medicine | Nucleoside derivatives |
| US5556948A (en) | 1993-01-22 | 1996-09-17 | Mitsubishi Chemical Corporation | Phospholipid derivatized with PEG bifunctional linker and liposome containing it |
| EP0425235B1 (en) | 1989-10-25 | 1996-09-25 | Immunogen Inc | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| US5565350A (en) | 1993-12-09 | 1996-10-15 | Thomas Jefferson University | Compounds and methods for site directed mutations in eukaryotic cells |
| US5567811A (en) | 1990-05-03 | 1996-10-22 | Amersham International Plc | Phosphoramidite derivatives, their preparation and the use thereof in the incorporation of reporter groups on synthetic oligonucleotides |
| US5569825A (en) | 1990-08-29 | 1996-10-29 | Genpharm International | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| WO1996034096A1 (en) | 1995-04-28 | 1996-10-31 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO1996033735A1 (en) | 1995-04-27 | 1996-10-31 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| US5576427A (en) | 1993-03-30 | 1996-11-19 | Sterling Winthrop, Inc. | Acyclic nucleoside analogs and oligonucleotide sequences containing them |
| US5578718A (en) | 1990-01-11 | 1996-11-26 | Isis Pharmaceuticals, Inc. | Thiol-derivatized nucleosides |
| US5580575A (en) | 1989-12-22 | 1996-12-03 | Imarx Pharmaceutical Corp. | Therapeutic drug delivery systems |
| US5580731A (en) | 1994-08-25 | 1996-12-03 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5583020A (en) | 1992-11-24 | 1996-12-10 | Ribozyme Pharmaceuticals, Inc. | Permeability enhancers for negatively charged polynucleotides |
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| US5585089A (en) | 1988-12-28 | 1996-12-17 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| US5587371A (en) | 1992-01-21 | 1996-12-24 | Pharmacyclics, Inc. | Texaphyrin-oligonucleotide conjugates |
| US5591722A (en) | 1989-09-15 | 1997-01-07 | Southern Research Institute | 2'-deoxy-4'-thioribonucleosides and their antiviral activity |
| US5591721A (en) | 1994-10-25 | 1997-01-07 | Hybridon, Inc. | Method of down-regulating gene expression |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5595726A (en) | 1992-01-21 | 1997-01-21 | Pharmacyclics, Inc. | Chromophore probe for detection of nucleic acid |
| US5595756A (en) | 1993-12-22 | 1997-01-21 | Inex Pharmaceuticals Corporation | Liposomal compositions for enhanced retention of bioactive agents |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5597909A (en) | 1994-08-25 | 1997-01-28 | Chiron Corporation | Polynucleotide reagents containing modified deoxyribose moieties, and associated methods of synthesis and use |
| US5599923A (en) | 1989-03-06 | 1997-02-04 | Board Of Regents, University Of Tx | Texaphyrin metal complexes having improved functionalization |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5606040A (en) | 1987-10-30 | 1997-02-25 | American Cyanamid Company | Antitumor and antibacterial substituted disulfide derivatives prepared from compounds possessing a methyl-trithio group |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5610300A (en) | 1992-07-01 | 1997-03-11 | Ciba-Geigy Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5623065A (en) | 1990-08-13 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Gapped 2' modified oligonucleotides |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US5625126A (en) | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5627053A (en) | 1994-03-29 | 1997-05-06 | Ribozyme Pharmaceuticals, Inc. | 2'deoxy-2'-alkylnucleotide containing nucleic acid |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5639873A (en) | 1992-02-05 | 1997-06-17 | Centre National De La Recherche Scientifique (Cnrs) | Oligothionucleotides |
| US5641870A (en) | 1995-04-20 | 1997-06-24 | Genentech, Inc. | Low pH hydrophobic interaction chromatography for antibody purification |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US5646265A (en) | 1990-01-11 | 1997-07-08 | Isis Pharmceuticals, Inc. | Process for the preparation of 2'-O-alkyl purine phosphoramidites |
| US5645985A (en) | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| US5652356A (en) | 1995-08-17 | 1997-07-29 | Hybridon, Inc. | Inverted chimeric and hybrid oligonucleotides |
| US5652355A (en) | 1992-07-23 | 1997-07-29 | Worcester Foundation For Experimental Biology | Hybrid oligonucleotide phosphorothioates |
| US5658873A (en) | 1993-04-10 | 1997-08-19 | Degussa Aktiengesellschaft | Coated sodium percarbonate particles, a process for their production and detergent, cleaning and bleaching compositions containing them |
| WO1997030087A1 (en) | 1996-02-16 | 1997-08-21 | Glaxo Group Limited | Preparation of glycosylated antibodies |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5663149A (en) | 1994-12-13 | 1997-09-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide heterocyclic and halophenyl amides |
| US5663312A (en) | 1993-03-31 | 1997-09-02 | Sanofi | Oligonucleotide dimers with amide linkages replacing phosphodiester linkages |
| WO1997033551A2 (en) | 1996-03-15 | 1997-09-18 | Millennium Pharmaceuticals | Compositions and methods for the diagnosis, prevention, and treatment of neoplastic cell growth and proliferation |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5677439A (en) | 1990-08-03 | 1997-10-14 | Sanofi | Oligonucleotide analogues containing phosphate diester linkage substitutes, compositions thereof, and precursor dinucleotide analogues |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5688941A (en) | 1990-07-27 | 1997-11-18 | Isis Pharmaceuticals, Inc. | Methods of making conjugated 4' desmethyl nucleoside analog compounds |
| US5700922A (en) | 1991-12-24 | 1997-12-23 | Isis Pharmaceuticals, Inc. | PNA-DNA-PNA chimeric macromolecules |
| US5712374A (en) | 1995-06-07 | 1998-01-27 | American Cyanamid Company | Method for the preparation of substantiallly monomeric calicheamicin derivative/carrier conjugates |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5739116A (en) | 1994-06-03 | 1998-04-14 | American Cyanamid Company | Enediyne derivatives useful for the synthesis of conjugates of methyltrithio antitumor agents |
| US5750692A (en) | 1990-01-11 | 1998-05-12 | Isis Pharmaceuticals, Inc. | Synthesis of 3-deazapurines |
| WO1998024893A2 (en) | 1996-12-03 | 1998-06-11 | Abgenix, Inc. | TRANSGENIC MAMMALS HAVING HUMAN IG LOCI INCLUDING PLURAL VH AND Vλ REGIONS AND ANTIBODIES PRODUCED THEREFROM |
| US5770701A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Process for preparing targeted forms of methyltrithio antitumor agents |
| US5780588A (en) | 1993-01-26 | 1998-07-14 | Arizona Board Of Regents | Elucidation and synthesis of selected pentapeptides |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5792608A (en) | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5792747A (en) | 1995-01-24 | 1998-08-11 | The Administrators Of The Tulane Educational Fund | Highly potent agonists of growth hormone releasing hormone |
| WO1998039352A1 (en) | 1997-03-07 | 1998-09-11 | Takeshi Imanishi | Novel bicyclonucleoside and oligonucleotide analogues |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| US5830653A (en) | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| WO1998058964A1 (en) | 1997-06-24 | 1998-12-30 | Genentech, Inc. | Methods and compositions for galactosylated glycoproteins |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| WO1999014226A2 (en) | 1997-09-12 | 1999-03-25 | Exiqon A/S | Bi- and tri-cyclic nucleoside, nucleotide and oligonucleotide analogues |
| WO1999022764A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| WO1999051642A1 (en) | 1998-04-02 | 1999-10-14 | Genentech, Inc. | Antibody variants and fragments thereof |
| US6004940A (en) | 1992-07-17 | 1999-12-21 | Dana-Farber Cancer Institute, Inc. | Intracellular targeting of endogenous proteins |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO2000042072A2 (en) | 1999-01-15 | 2000-07-20 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| US6136958A (en) * | 1996-09-30 | 2000-10-24 | Genentech, Inc. | Antibodies to vertebrate smoothened proteins |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| WO2001029246A1 (en) | 1999-10-19 | 2001-04-26 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing polypeptide |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| US20020025313A1 (en) | 1992-07-27 | 2002-02-28 | Micklus Michael J. | Targeting of liposomes to the blood-brain barrier |
| US20020038086A1 (en) | 2000-07-27 | 2002-03-28 | Hynynen Kullervo H. | Blood-brain barrier opening |
| WO2002031140A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions |
| US20020065259A1 (en) | 2000-08-30 | 2002-05-30 | Schatzberg Alan F. | Glucocorticoid blocking agents for increasing blood-brain barrier permeability |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| US20020164328A1 (en) | 2000-10-06 | 2002-11-07 | Toyohide Shinkawa | Process for purifying antibody |
| WO2002088172A2 (en) | 2001-04-30 | 2002-11-07 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| WO2003011878A2 (en) | 2001-08-03 | 2003-02-13 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| US20030073713A1 (en) | 2000-10-30 | 2003-04-17 | Schoenhard Grant L. | Inhibitors of ABC drug transporters at the blood-brain barrier |
| US20030083299A1 (en) | 2000-11-04 | 2003-05-01 | Ferguson Ian A. | Non-invasive delivery of polypeptides through the blood-brain barrier |
| US20030104402A1 (en) | 2001-01-23 | 2003-06-05 | University Of Rochester | Methods of producing or identifying intrabodies in eukaryotic cells |
| US20030115614A1 (en) | 2000-10-06 | 2003-06-19 | Yutaka Kanda | Antibody composition-producing cell |
| US20030129186A1 (en) | 2001-07-25 | 2003-07-10 | Biomarin Pharmaceutical Inc. | Compositions and methods for modulating blood-brain barrier transport |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US20030157108A1 (en) | 2001-10-25 | 2003-08-21 | Genentech, Inc. | Glycoprotein compositions |
| US20030162695A1 (en) | 2002-02-27 | 2003-08-28 | Schatzberg Alan F. | Glucocorticoid blocking agents for increasing blood-brain barrier permeability |
| WO2003077945A1 (en) | 2002-03-14 | 2003-09-25 | Medical Research Council | Intracellular antibodies |
| WO2003084570A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | DRUG CONTAINING ANTIBODY COMPOSITION APPROPRIATE FOR PATIENT SUFFERING FROM FcϜRIIIa POLYMORPHISM |
| WO2003085119A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | METHOD OF ENHANCING ACTIVITY OF ANTIBODY COMPOSITION OF BINDING TO FcϜ RECEPTOR IIIa |
| WO2003085107A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Cells with modified genome |
| US6656730B1 (en) | 1999-06-15 | 2003-12-02 | Isis Pharmaceuticals, Inc. | Oligonucleotides conjugated to protein-binding drugs |
| EP1391213A1 (en) | 2002-08-21 | 2004-02-25 | Boehringer Ingelheim International GmbH | Compositions and methods for treating cancer using maytansinoid CD44 antibody immunoconjugates and chemotherapeutic agents |
| US6703019B1 (en) | 1991-04-30 | 2004-03-09 | Bernard Malfroy-Camine | Cationized antibodies against intracellular proteins |
| US20040093621A1 (en) | 2001-12-25 | 2004-05-13 | Kyowa Hakko Kogyo Co., Ltd | Antibody composition which specifically binds to CD20 |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US20040110282A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells in which activity of the protein involved in transportation of GDP-fucose is reduced or lost |
| US20040109865A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition-containing medicament |
| US20040132140A1 (en) | 2002-04-09 | 2004-07-08 | Kyowa Hakko Kogyo Co., Ltd. | Production process for antibody composition |
| WO2004056312A2 (en) | 2002-12-16 | 2004-07-08 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| US20040131692A1 (en) | 2001-05-05 | 2004-07-08 | Joerg Kreuter | Nanoparticles made of protein with coupled apolipoprotein e for penetration of the blood-brain barrier and methods for the production thereof |
| US20040204354A1 (en) | 2002-12-03 | 2004-10-14 | Thomas Nelson | Artificial low-density lipoprotein carriers for transport of substances across the blood-brain barrier |
| WO2004092219A2 (en) | 2003-04-10 | 2004-10-28 | Protein Design Labs, Inc | Alteration of fcrn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20050014934A1 (en) | 2002-10-15 | 2005-01-20 | Hinton Paul R. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20050026229A1 (en) | 1997-03-10 | 2005-02-03 | The Regents Of The University Of California | PSCA antibodies and hybridomas producing them |
| WO2005035586A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Fused protein composition |
| WO2005035778A1 (en) | 2003-10-09 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | PROCESS FOR PRODUCING ANTIBODY COMPOSITION BY USING RNA INHIBITING THE FUNCTION OF α1,6-FUCOSYLTRANSFERASE |
| US20050089473A1 (en) | 2003-09-10 | 2005-04-28 | Cedars-Sinai Medical Center | Potassium channel mediated delivery of agents through the blood-brain barrier |
| US20050100546A1 (en) | 1997-05-05 | 2005-05-12 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| US20050123546A1 (en) | 2003-11-05 | 2005-06-09 | Glycart Biotechnology Ag | Antigen binding molecules with increased Fc receptor binding affinity and effector function |
| WO2005053742A1 (en) | 2003-12-04 | 2005-06-16 | Kyowa Hakko Kogyo Co., Ltd. | Medicine containing antibody composition |
| US6919436B2 (en) | 1996-08-30 | 2005-07-19 | Upfront Chromatography A/S | Isolation of proteins |
| US20050287149A1 (en) | 2003-05-21 | 2005-12-29 | Medarex, Inc. | Human monoclonal antibodies against bacillus anthracis protective antigen |
| US6982321B2 (en) | 1986-03-27 | 2006-01-03 | Medical Research Council | Altered antibodies |
| US20060059575A1 (en) | 1999-03-30 | 2006-03-16 | Japan Tobacco, Inc. | Method for preparing monoclonal antibody |
| US7078492B2 (en) | 2001-05-16 | 2006-07-18 | Abgenix, Inc. | Human antipneumococcal antibodies from non-human animals |
| US7087409B2 (en) | 1997-12-05 | 2006-08-08 | The Scripps Research Institute | Humanization of murine antibody |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| US20060258841A1 (en) | 2003-01-17 | 2006-11-16 | Josef Michl | Pancreatic cancer associated antigen, antibody thereto, and diagnostic and treatment methods |
| US7153507B2 (en) | 2001-08-23 | 2006-12-26 | Genmab A/S | Human antibodies specific for interleukin 15 (IL-15) |
| US7189826B2 (en) | 1997-11-24 | 2007-03-13 | Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5990281A (en) | 1996-09-30 | 1999-11-23 | Genentech, Inc. | Vertebrate smoothened proteins |
| WO2007059157A1 (en) | 2005-11-14 | 2007-05-24 | Genentech, Inc. | Bisamide inhibitors of hedgehog signaling |
| JP5974012B2 (en) | 2010-10-05 | 2016-08-23 | ジェネンテック, インコーポレイテッド | Mutant smoothened and method of using the same |
-
2010
- 2010-09-02 CA CA2772715A patent/CA2772715C/en not_active Expired - Fee Related
- 2010-09-02 ES ES10751758.3T patent/ES2599076T3/en active Active
- 2010-09-02 JP JP2012528067A patent/JP5887270B2/en not_active Expired - Fee Related
- 2010-09-02 DK DK10751758.3T patent/DK2473522T3/en active
- 2010-09-02 AU AU2010289400A patent/AU2010289400B2/en not_active Ceased
- 2010-09-02 US US13/394,069 patent/US9321823B2/en active Active
- 2010-09-02 EP EP10751758.3A patent/EP2473522B1/en not_active Not-in-force
- 2010-09-02 WO PCT/US2010/047739 patent/WO2011028950A1/en active Application Filing
-
2015
- 2015-09-30 JP JP2015194348A patent/JP6279527B2/en not_active Expired - Fee Related
-
2016
- 2016-04-15 US US15/130,223 patent/US9910050B2/en not_active Expired - Fee Related
-
2018
- 2018-01-16 JP JP2018005035A patent/JP2018061516A/en active Pending
Patent Citations (387)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3720760A (en) | 1968-09-06 | 1973-03-13 | Pharmacia Ab | Method for determining the presence of reagin-immunoglobulins(reagin-ig)directed against certain allergens,in aqueous samples |
| US3720760B1 (en) | 1968-09-06 | 1984-02-07 | Pharmacia Ab | |
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US3896111A (en) | 1973-02-20 | 1975-07-22 | Research Corp | Ansa macrolides |
| US4151042A (en) | 1977-03-31 | 1979-04-24 | Takeda Chemical Industries, Ltd. | Method for producing maytansinol and its derivatives |
| US4137230A (en) | 1977-11-14 | 1979-01-30 | Takeda Chemical Industries, Ltd. | Method for the production of maytansinoids |
| USRE30985E (en) | 1978-01-01 | 1982-06-29 | Serum-free cell culture media | |
| EP0003089A1 (en) | 1978-01-06 | 1979-07-25 | Bernard David | Drier for silkscreen printed sheets |
| US4265814A (en) | 1978-03-24 | 1981-05-05 | Takeda Chemical Industries | Matansinol 3-n-hexadecanoate |
| US4307016A (en) | 1978-03-24 | 1981-12-22 | Takeda Chemical Industries, Ltd. | Demethyl maytansinoids |
| US4361650A (en) | 1978-03-24 | 1982-11-30 | Takeda Chemical Industries, Ltd. | Fermentation process of preparing demethyl maytansinoids |
| US4248870A (en) | 1978-10-27 | 1981-02-03 | Takeda Chemical Industries, Ltd. | Maytansinoids and use |
| US4260608A (en) | 1978-11-14 | 1981-04-07 | Takeda Chemical Industries, Ltd. | Maytansinoids, pharmaceutical compositions thereof and methods of use thereof |
| US4256746A (en) | 1978-11-14 | 1981-03-17 | Takeda Chemical Industries | Dechloromaytansinoids, their pharmaceutical compositions and method of use |
| US4294757A (en) | 1979-01-31 | 1981-10-13 | Takeda Chemical Industries, Ltd | 20-O-Acylmaytansinoids |
| US4322348A (en) | 1979-06-05 | 1982-03-30 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| US4317821A (en) | 1979-06-08 | 1982-03-02 | Takeda Chemical Industries, Ltd. | Maytansinoids, their use and pharmaceutical compositions thereof |
| US4308269A (en) | 1979-06-11 | 1981-12-29 | Takeda Chemical Industries, Ltd. | Maytansinoids, pharmaceutical compositions thereof and method of use thereof |
| US4308268A (en) | 1979-06-11 | 1981-12-29 | Takeda Chemical Industries, Ltd. | Maytansinoids, pharmaceutical compositions thereof and method of use thereof |
| US4309428A (en) | 1979-07-30 | 1982-01-05 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| US4331598A (en) | 1979-09-19 | 1982-05-25 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| US4362663A (en) | 1979-09-21 | 1982-12-07 | Takeda Chemical Industries, Ltd. | Maytansinoid compound |
| US4364866A (en) | 1979-09-21 | 1982-12-21 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| US4371533A (en) | 1980-10-08 | 1983-02-01 | Takeda Chemical Industries, Ltd. | 4,5-Deoxymaytansinoids, their use and pharmaceutical compositions thereof |
| US4450254A (en) | 1980-11-03 | 1984-05-22 | Standard Oil Company | Impact improvement of high nitrile resins |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4419446A (en) | 1980-12-31 | 1983-12-06 | The United States Of America As Represented By The Department Of Health And Human Services | Recombinant DNA process utilizing a papilloma virus DNA as a vector |
| US4313946A (en) | 1981-01-27 | 1982-02-02 | The United States Of America As Represented By The Secretary Of Agriculture | Chemotherapeutically active maytansinoids from Trewia nudiflora |
| US4315929A (en) | 1981-01-27 | 1982-02-16 | The United States Of America As Represented By The Secretary Of Agriculture | Method of controlling the European corn borer with trewiasine |
| US4424219A (en) | 1981-05-20 | 1984-01-03 | Takeda Chemical Industries, Ltd. | 9-Thiomaytansinoids and their pharmaceutical compositions and use |
| US4534899A (en) | 1981-07-20 | 1985-08-13 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US4426330A (en) | 1981-07-20 | 1984-01-17 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| EP0073657A1 (en) | 1981-08-31 | 1983-03-09 | Genentech, Inc. | Preparation of hepatitis B surface antigen in yeast |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US4667025A (en) | 1982-08-09 | 1987-05-19 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4789737A (en) | 1982-08-09 | 1988-12-06 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives and production thereof |
| US4601978A (en) | 1982-11-24 | 1986-07-22 | The Regents Of The University Of California | Mammalian metallothionein promoter system |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4835263A (en) | 1983-01-27 | 1989-05-30 | Centre National De La Recherche Scientifique | Novel compounds containing an oligonucleotide sequence bonded to an intercalating agent, a process for their synthesis and their use |
| US4605735A (en) | 1983-02-14 | 1986-08-12 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4948882A (en) | 1983-02-22 | 1990-08-14 | Syngene, Inc. | Single-stranded labelled oligonucleotides, reactive monomers and methods of synthesis |
| US5541313A (en) | 1983-02-22 | 1996-07-30 | Molecular Biosystems, Inc. | Single-stranded labelled oligonucleotides of preselected sequence |
| US4824941A (en) | 1983-03-10 | 1989-04-25 | Julian Gordon | Specific antibody to the native form of 2'5'-oligonucleotides, the method of preparation and the use as reagents in immunoassays or for binding 2'5'-oligonucleotides in biological systems |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| DD266710A3 (en) | 1983-06-06 | 1989-04-12 | Ve Forschungszentrum Biotechnologie | Process for the biotechnical production of alkaline phosphatase |
| US4587044A (en) | 1983-09-01 | 1986-05-06 | The Johns Hopkins University | Linkage of proteins to nucleic acids |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US5118800A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | Oligonucleotides possessing a primary amino group in the terminal nucleotide |
| US5118802A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | DNA-reporter conjugates linked via the 2' or 5'-primary amino group of the 5'-terminal nucleoside |
| US4965199A (en) | 1984-04-20 | 1990-10-23 | Genentech, Inc. | Preparation of functional human factor VIII in mammalian cells using methotrexate based selection |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US4981957A (en) | 1984-07-19 | 1991-01-01 | Centre National De La Recherche Scientifique | Oligonucleotides with modified phosphate and modified carbohydrate moieties at the respective chain termini |
| US5258506A (en) | 1984-10-16 | 1993-11-02 | Chiron Corporation | Photolabile reagents for incorporation into oligonucleotide chains |
| US5578717A (en) | 1984-10-16 | 1996-11-26 | Chiron Corporation | Nucleotides for introducing selectably cleavable and/or abasic sites into oligonucleotides |
| US5552538A (en) | 1984-10-16 | 1996-09-03 | Chiron Corporation | Oligonucleotides with cleavable sites |
| US5545730A (en) | 1984-10-16 | 1996-08-13 | Chiron Corporation | Multifunctional nucleic acid monomer |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| EP0183070A2 (en) | 1984-10-30 | 1986-06-04 | Phillips Petroleum Company | Transformation of yeasts of the genus pichia |
| US4828979A (en) | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| US4845205A (en) | 1985-01-08 | 1989-07-04 | Institut Pasteur | 2,N6 -disubstituted and 2,N6 -trisubstituted adenosine-3'-phosphoramidites |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US4737456A (en) | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| US4762779A (en) | 1985-06-13 | 1988-08-09 | Amgen Inc. | Compositions and methods for functionalizing nucleic acids |
| WO1987000195A1 (en) | 1985-06-28 | 1987-01-15 | Celltech Limited | Animal cell culture |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US4970198A (en) | 1985-10-17 | 1990-11-13 | American Cyanamid Company | Antitumor antibiotics (LL-E33288 complex) |
| US5317098A (en) | 1986-03-17 | 1994-05-31 | Hiroaki Shizuya | Non-radioisotope tagging of fragments |
| US6982321B2 (en) | 1986-03-27 | 2006-01-03 | Medical Research Council | Altered antibodies |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| EP0244234A2 (en) | 1986-04-30 | 1987-11-04 | Alko Group Ltd. | Transformation of trichoderma |
| US4876335A (en) | 1986-06-30 | 1989-10-24 | Wakunaga Seiyaku Kabushiki Kaisha | Poly-labelled oligonucleotide derivative |
| US5013830A (en) | 1986-09-08 | 1991-05-07 | Ajinomoto Co., Inc. | Compounds for the cleavage at a specific position of RNA, oligomers employed for the formation of said compounds, and starting materials for the synthesis of said oligomers |
| US5500362A (en) | 1987-01-08 | 1996-03-19 | Xoma Corporation | Chimeric antibody with specificity to human B cell surface antigen |
| US5079233A (en) | 1987-01-30 | 1992-01-07 | American Cyanamid Company | N-acyl derivatives of the LL-E33288 antitumor antibiotics, composition and methods for using the same |
| US5648260A (en) | 1987-03-18 | 1997-07-15 | Scotgen Biopharmaceuticals Incorporated | DNA encoding antibodies with altered effector functions |
| US5624821A (en) | 1987-03-18 | 1997-04-29 | Scotgen Biopharmaceuticals Incorporated | Antibodies with altered effector functions |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5286717A (en) | 1987-03-25 | 1994-02-15 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| US5552540A (en) | 1987-06-24 | 1996-09-03 | Howard Florey Institute Of Experimental Physiology And Medicine | Nucleoside derivatives |
| US5004697A (en) | 1987-08-17 | 1991-04-02 | Univ. Of Ca | Cationized antibodies for delivery through the blood-brain barrier |
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5525465A (en) | 1987-10-28 | 1996-06-11 | Howard Florey Institute Of Experimental Physiology And Medicine | Oligonucleotide-polyamide conjugates and methods of production and applications of the same |
| US5770701A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Process for preparing targeted forms of methyltrithio antitumor agents |
| US5770710A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Antitumor and antibacterial substituted disulfide derivatives prepared from compounds possessing a methlytrithio group |
| US5606040A (en) | 1987-10-30 | 1997-02-25 | American Cyanamid Company | Antitumor and antibacterial substituted disulfide derivatives prepared from compounds possessing a methyl-trithio group |
| US5112963A (en) | 1987-11-12 | 1992-05-12 | Max-Planck-Gesellschaft Zur Foerderung Der Wissenschaften E.V. | Modified oligonucleotides |
| US5283187A (en) | 1987-11-17 | 1994-02-01 | Brown University Research Foundation | Cell culture-containing tubular capsule produced by co-extrusion |
| US4892538A (en) | 1987-11-17 | 1990-01-09 | Brown University Research Foundation | In vivo delivery of neurotransmitters by implanted, encapsulated cells |
| US5491133A (en) | 1987-11-30 | 1996-02-13 | University Of Iowa Research Foundation | Methods for blocking the expression of specifically targeted genes |
| US5403711A (en) | 1987-11-30 | 1995-04-04 | University Of Iowa Research Foundation | Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved |
| US5082830A (en) | 1988-02-26 | 1992-01-21 | Enzo Biochem, Inc. | End labeled nucleotide probe |
| US4975278A (en) | 1988-02-26 | 1990-12-04 | Bristol-Myers Company | Antibody-enzyme conjugates in combination with prodrugs for the delivery of cytotoxic agents to tumor cells |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5453496A (en) | 1988-05-26 | 1995-09-26 | University Patents, Inc. | Polynucleotide phosphorodithioate |
| US5109124A (en) | 1988-06-01 | 1992-04-28 | Biogen, Inc. | Nucleic acid probe linked to a label having a terminal cysteine |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5262536A (en) | 1988-09-15 | 1993-11-16 | E. I. Du Pont De Nemours And Company | Reagents for the preparation of 5'-tagged oligonucleotides |
| US5053394A (en) | 1988-09-21 | 1991-10-01 | American Cyanamid Company | Targeted forms of methyltrithio antitumor agents |
| US5565555A (en) | 1988-09-23 | 1996-10-15 | Gilead Sciences, Inc. | Nucleoside hydrogen phosphonodithioate diesters and activated phosphonodithioate analogues |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| WO1990003430A1 (en) | 1988-09-23 | 1990-04-05 | Cetus Corporation | Cell culture medium for enhanced cell growth, culture longevity and product expression |
| US5545807A (en) | 1988-10-12 | 1996-08-13 | The Babraham Institute | Production of antibodies from transgenic animals |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| US5512439A (en) | 1988-11-21 | 1996-04-30 | Dynal As | Oligonucleotide-linked magnetic particles and uses thereof |
| US5585089A (en) | 1988-12-28 | 1996-12-17 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5693762A (en) | 1988-12-28 | 1997-12-02 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| WO1990010048A1 (en) | 1989-02-27 | 1990-09-07 | Kerr-Mcgee Corporation | Solvent extraction process |
| US5599923A (en) | 1989-03-06 | 1997-02-04 | Board Of Regents, University Of Tx | Texaphyrin metal complexes having improved functionalization |
| WO1990010448A2 (en) | 1989-03-07 | 1990-09-20 | Genentech, Inc. | Covalent conjugates of lipid and oligonucleotide |
| US5354844A (en) | 1989-03-16 | 1994-10-11 | Boehringer Ingelheim International Gmbh | Protein-polycation conjugates |
| US5416016A (en) | 1989-04-03 | 1995-05-16 | Purdue Research Foundation | Method for enhancing transmembrane transport of exogenous molecules |
| US5108921A (en) | 1989-04-03 | 1992-04-28 | Purdue Research Foundation | Method for enhanced transmembrane transport of exogenous molecules |
| WO1990013646A1 (en) | 1989-04-28 | 1990-11-15 | Transgene S.A. | Application of novel dna fragments as a coding sequence for a signal peptide for the secretion of mature proteins by recombinant yeast, expression cassettes, transformed yeasts and corresponding process for the preparation of proteins |
| WO1990013641A1 (en) | 1989-05-10 | 1990-11-15 | Sloan-Kettering Institute For Cancer Research | Stably transformed eucaryotic cells comprising a foreign transcribable dna under the control of a pol iii promoter |
| US5391723A (en) | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US5256775A (en) | 1989-06-05 | 1993-10-26 | Gilead Sciences, Inc. | Exonuclease-resistant oligonucleotides |
| US4958013A (en) | 1989-06-06 | 1990-09-18 | Northwestern University | Cholesteryl modified oligonucleotides |
| EP0402226A1 (en) | 1989-06-06 | 1990-12-12 | Institut National De La Recherche Agronomique | Transformation vectors for yeast yarrowia |
| US5416203A (en) | 1989-06-06 | 1995-05-16 | Northwestern University | Steroid modified oligonucleotides |
| US5227170A (en) | 1989-06-22 | 1993-07-13 | Vestar, Inc. | Encapsulation process |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| WO1991000360A1 (en) | 1989-06-29 | 1991-01-10 | Medarex, Inc. | Bispecific reagents for aids therapy |
| US5451463A (en) | 1989-08-28 | 1995-09-19 | Clontech Laboratories, Inc. | Non-nucleoside 1,3-diol reagents for labeling synthetic oligonucleotides |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5254469A (en) | 1989-09-12 | 1993-10-19 | Eastman Kodak Company | Oligonucleotide-enzyme conjugate that can be used as a probe in hybridization assays and polymerase chain reaction procedures |
| US5591722A (en) | 1989-09-15 | 1997-01-07 | Southern Research Institute | 2'-deoxy-4'-thioribonucleosides and their antiviral activity |
| WO1991004753A1 (en) | 1989-10-02 | 1991-04-18 | Cetus Corporation | Conjugates of antisense oligonucleotides and therapeutic uses thereof |
| US5213804A (en) | 1989-10-20 | 1993-05-25 | Liposome Technology, Inc. | Solid tumor treatment method and composition |
| US5527528A (en) | 1989-10-20 | 1996-06-18 | Sequus Pharmaceuticals, Inc. | Solid-tumor treatment method |
| US5356633A (en) | 1989-10-20 | 1994-10-18 | Liposome Technology, Inc. | Method of treatment of inflamed tissues |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5527899A (en) | 1989-10-23 | 1996-06-18 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| WO1991006629A1 (en) | 1989-10-24 | 1991-05-16 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5466786B1 (en) | 1989-10-24 | 1998-04-07 | Gilead Sciences | 2' Modified nucleoside and nucleotide compounds |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5466786A (en) | 1989-10-24 | 1995-11-14 | Gilead Sciences | 2'modified nucleoside and nucleotide compounds |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| EP0425235B1 (en) | 1989-10-25 | 1996-09-25 | Immunogen Inc | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5416064A (en) | 1989-10-25 | 1995-05-16 | Immunogen, Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US6417429B1 (en) | 1989-10-27 | 2002-07-09 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US5292873A (en) | 1989-11-29 | 1994-03-08 | The Research Foundation Of State University Of New York | Nucleic acids labeled with naphthoquinone probe |
| US5455233A (en) | 1989-11-30 | 1995-10-03 | University Of North Carolina | Oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5580575A (en) | 1989-12-22 | 1996-12-03 | Imarx Pharmaceutical Corp. | Therapeutic drug delivery systems |
| US5469854A (en) | 1989-12-22 | 1995-11-28 | Imarx Pharmaceutical Corp. | Methods of preparing gas-filled liposomes |
| US5486603A (en) | 1990-01-08 | 1996-01-23 | Gilead Sciences, Inc. | Oligonucleotide having enhanced binding affinity |
| US5646265A (en) | 1990-01-11 | 1997-07-08 | Isis Pharmceuticals, Inc. | Process for the preparation of 2'-O-alkyl purine phosphoramidites |
| US5587469A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides containing N-2 substituted purines |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5578718A (en) | 1990-01-11 | 1996-11-26 | Isis Pharmaceuticals, Inc. | Thiol-derivatized nucleosides |
| US5750692A (en) | 1990-01-11 | 1998-05-12 | Isis Pharmaceuticals, Inc. | Synthesis of 3-deazapurines |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO1991010741A1 (en) | 1990-01-12 | 1991-07-25 | Cell Genesys, Inc. | Generation of xenogeneic antibodies |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5220007A (en) | 1990-02-15 | 1993-06-15 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5366878A (en) | 1990-02-15 | 1994-11-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of RNA and production of encoded polypeptides |
| US5149797A (en) | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5414077A (en) | 1990-02-20 | 1995-05-09 | Gilead Sciences | Non-nucleoside linkers for convenient attachment of labels to oligonucleotides using standard synthetic methods |
| US5214136A (en) | 1990-02-20 | 1993-05-25 | Gilead Sciences, Inc. | Anthraquinone-derivatives oligonucleotides |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5541306A (en) | 1990-03-08 | 1996-07-30 | Worcester Foundation For Biomedical Research | Aminoalkylphosphotriester oligonucleotide derivatives |
| US5563253A (en) | 1990-03-08 | 1996-10-08 | Worcester Foundation For Biomedical Research | Linear aminoalkylphosphoramidate oligonucleotide derivatives |
| US5536821A (en) | 1990-03-08 | 1996-07-16 | Worcester Foundation For Biomedical Research | Aminoalkylphosphorothioamidate oligonucleotide deratives |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5459127A (en) | 1990-04-19 | 1995-10-17 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| US5506206A (en) | 1990-04-23 | 1996-04-09 | Alkermes, Inc. | Increasing blood-brain barrier permeability with permeabilizer peptides |
| US5112596A (en) | 1990-04-23 | 1992-05-12 | Alkermes, Inc. | Method for increasing blood-brain barrier permeability by administering a bradykinin agonist of blood-brain barrier permeability |
| US5268164A (en) | 1990-04-23 | 1993-12-07 | Alkermes, Inc. | Increasing blood-brain barrier permeability with permeabilizer peptides |
| US5567811A (en) | 1990-05-03 | 1996-10-22 | Amersham International Plc | Phosphoramidite derivatives, their preparation and the use thereof in the incorporation of reporter groups on synthetic oligonucleotides |
| US5514785A (en) | 1990-05-11 | 1996-05-07 | Becton Dickinson And Company | Solid supports for nucleic acid hybridization assays |
| WO1992000373A1 (en) | 1990-06-29 | 1992-01-09 | Biosource Genetics Corporation | Melanin production by transformed microorganisms |
| US5218105A (en) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5688941A (en) | 1990-07-27 | 1997-11-18 | Isis Pharmaceuticals, Inc. | Methods of making conjugated 4' desmethyl nucleoside analog compounds |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5138045A (en) | 1990-07-27 | 1992-08-11 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5677439A (en) | 1990-08-03 | 1997-10-14 | Sanofi | Oligonucleotide analogues containing phosphate diester linkage substitutes, compositions thereof, and precursor dinucleotide analogues |
| US5567810A (en) | 1990-08-03 | 1996-10-22 | Sterling Drug, Inc. | Nuclease resistant compounds |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| US5623065A (en) | 1990-08-13 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Gapped 2' modified oligonucleotides |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5512667A (en) | 1990-08-28 | 1996-04-30 | Reed; Michael W. | Trifunctional intermediates for preparing 3'-tailed oligonucleotides |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5625126A (en) | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5569825A (en) | 1990-08-29 | 1996-10-29 | Genpharm International | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5510475A (en) | 1990-11-08 | 1996-04-23 | Hybridon, Inc. | Oligonucleotide multiple reporter precursors |
| WO1992009690A2 (en) | 1990-12-03 | 1992-06-11 | Genentech, Inc. | Enrichment method for variant proteins with altered binding properties |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5686416A (en) | 1991-04-23 | 1997-11-11 | Alkermes, Inc. | Increasing blood-brain barrier permeability with permeabilizer peptides |
| US6703019B1 (en) | 1991-04-30 | 2004-03-09 | Bernard Malfroy-Camine | Cationized antibodies against intracellular proteins |
| US5264221A (en) | 1991-05-23 | 1993-11-23 | Mitsubishi Kasei Corporation | Drug-containing protein-bonded liposome |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5821337A (en) | 1991-06-14 | 1998-10-13 | Genentech, Inc. | Immunoglobulin variants |
| WO1993001161A1 (en) | 1991-07-11 | 1993-01-21 | Pfizer Limited | Process for preparing sertraline intermediates |
| US5371241A (en) | 1991-07-19 | 1994-12-06 | Pharmacia P-L Biochemicals Inc. | Fluorescein labelled phosphoramidites |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5362852A (en) | 1991-09-27 | 1994-11-08 | Pfizer Inc. | Modified peptide derivatives conjugated at 2-hydroxyethylamine moieties |
| US5521291A (en) | 1991-09-30 | 1996-05-28 | Boehringer Ingelheim International, Gmbh | Conjugates for introducing nucleic acid into higher eucaryotic cells |
| US5547932A (en) | 1991-09-30 | 1996-08-20 | Boehringer Ingelheim International Gmbh | Composition for introducing nucleic acid complexes into higher eucaryotic cells |
| US5587458A (en) | 1991-10-07 | 1996-12-24 | Aronex Pharmaceuticals, Inc. | Anti-erbB-2 antibodies, combinations thereof, and therapeutic and diagnostic uses thereof |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5393878A (en) | 1991-10-17 | 1995-02-28 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| US5319080A (en) | 1991-10-17 | 1994-06-07 | Ciba-Geigy Corporation | Bicyclic nucleosides, oligonucleotides, process for their preparation and intermediates |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5830653A (en) | 1991-11-26 | 1998-11-03 | Gilead Sciences, Inc. | Methods of using oligomers containing modified pyrimidines |
| US5645985A (en) | 1991-11-26 | 1997-07-08 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5792608A (en) | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| US5700922A (en) | 1991-12-24 | 1997-12-23 | Isis Pharmaceuticals, Inc. | PNA-DNA-PNA chimeric macromolecules |
| US5595726A (en) | 1992-01-21 | 1997-01-21 | Pharmacyclics, Inc. | Chromophore probe for detection of nucleic acid |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| US5587371A (en) | 1992-01-21 | 1996-12-24 | Pharmacyclics, Inc. | Texaphyrin-oligonucleotide conjugates |
| US5639873A (en) | 1992-02-05 | 1997-06-17 | Centre National De La Recherche Scientifique (Cnrs) | Oligothionucleotides |
| WO1993016185A2 (en) | 1992-02-06 | 1993-08-19 | Creative Biomolecules, Inc. | Biosynthetic binding protein for cancer marker |
| WO1993021232A1 (en) | 1992-04-10 | 1993-10-28 | Research Development Foundation | IMMUNOTOXINS DIRECTED AGAINST c-erbB-2 (HER-2/neu) RELATED SURFACE ANTIGENS |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| WO1993025673A1 (en) | 1992-06-04 | 1993-12-23 | The Regents Of The University Of California | In vivo gene therapy with intron-free sequence of interest |
| US5610300A (en) | 1992-07-01 | 1997-03-11 | Ciba-Geigy Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5700920A (en) | 1992-07-01 | 1997-12-23 | Novartis Corporation | Carbocyclic nucleosides containing bicyclic rings, oligonucleotides therefrom, process for their preparation, their use and intermediates |
| US5272250A (en) | 1992-07-10 | 1993-12-21 | Spielvogel Bernard F | Boronated phosphoramidate compounds |
| US6004940A (en) | 1992-07-17 | 1999-12-21 | Dana-Farber Cancer Institute, Inc. | Intracellular targeting of endogenous proteins |
| US6329173B1 (en) | 1992-07-17 | 2001-12-11 | Dana-Farber Cancer Institute, Inc. | Method of intracellular binding target molecules |
| WO1994002935A1 (en) | 1992-07-22 | 1994-02-03 | Sinvent A/S | Method and device for active noise reduction in a local area |
| US5652355A (en) | 1992-07-23 | 1997-07-29 | Worcester Foundation For Experimental Biology | Hybrid oligonucleotide phosphorothioates |
| US20020025313A1 (en) | 1992-07-27 | 2002-02-28 | Micklus Michael J. | Targeting of liposomes to the blood-brain barrier |
| WO1994004690A1 (en) | 1992-08-17 | 1994-03-03 | Genentech, Inc. | Bispecific immunoadhesins |
| WO1994011026A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Therapeutic application of chimeric and radiolabeled antibodies to human b lymphocyte restricted differentiation antigen for treatment of b cell lymphoma |
| US5583020A (en) | 1992-11-24 | 1996-12-10 | Ribozyme Pharmaceuticals, Inc. | Permeability enhancers for negatively charged polynucleotides |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| US5556948A (en) | 1993-01-22 | 1996-09-17 | Mitsubishi Chemical Corporation | Phospholipid derivatized with PEG bifunctional linker and liposome containing it |
| US5780588A (en) | 1993-01-26 | 1998-07-14 | Arizona Board Of Regents | Elucidation and synthesis of selected pentapeptides |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5395619A (en) | 1993-03-03 | 1995-03-07 | Liposome Technology, Inc. | Lipid-polymer conjugates and liposomes |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5576427A (en) | 1993-03-30 | 1996-11-19 | Sterling Winthrop, Inc. | Acyclic nucleoside analogs and oligonucleotide sequences containing them |
| US5663312A (en) | 1993-03-31 | 1997-09-02 | Sanofi | Oligonucleotide dimers with amide linkages replacing phosphodiester linkages |
| US5658873A (en) | 1993-04-10 | 1997-08-19 | Degussa Aktiengesellschaft | Coated sodium percarbonate particles, a process for their production and detergent, cleaning and bleaching compositions containing them |
| US5462854A (en) | 1993-04-19 | 1995-10-31 | Beckman Instruments, Inc. | Inverse linkage oligonucleotides for chemical and enzymatic processes |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5534259A (en) | 1993-07-08 | 1996-07-09 | Liposome Technology, Inc. | Polymer compound and coated particle composition |
| US5543158A (en) | 1993-07-23 | 1996-08-06 | Massachusetts Institute Of Technology | Biodegradable injectable nanoparticles |
| US5417978A (en) | 1993-07-29 | 1995-05-23 | Board Of Regents, The University Of Texas System | Liposomal antisense methyl phosphonate oligonucleotides and methods for their preparation and use |
| US6005096A (en) | 1993-09-17 | 1999-12-21 | Gilead Sciences, Inc. | Pyrimidine derivatives |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5763588A (en) | 1993-09-17 | 1998-06-09 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5565350A (en) | 1993-12-09 | 1996-10-15 | Thomas Jefferson University | Compounds and methods for site directed mutations in eukaryotic cells |
| US5446137A (en) | 1993-12-09 | 1995-08-29 | Syntex (U.S.A.) Inc. | Oligonucleotides containing 4'-substituted nucleotides |
| US5446137B1 (en) | 1993-12-09 | 1998-10-06 | Behringwerke Ag | Oligonucleotides containing 4'-substituted nucleotides |
| US5595756A (en) | 1993-12-22 | 1997-01-21 | Inex Pharmaceuticals Corporation | Liposomal compositions for enhanced retention of bioactive agents |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5599928A (en) | 1994-02-15 | 1997-02-04 | Pharmacyclics, Inc. | Texaphyrin compounds having improved functionalization |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5627053A (en) | 1994-03-29 | 1997-05-06 | Ribozyme Pharmaceuticals, Inc. | 2'deoxy-2'-alkylnucleotide containing nucleic acid |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5739116A (en) | 1994-06-03 | 1998-04-14 | American Cyanamid Company | Enediyne derivatives useful for the synthesis of conjugates of methyltrithio antitumor agents |
| US5773001A (en) | 1994-06-03 | 1998-06-30 | American Cyanamid Company | Conjugates of methyltrithio antitumor agents and intermediates for their synthesis |
| US5877296A (en) | 1994-06-03 | 1999-03-02 | American Cyanamid Company | Process for preparing conjugates of methyltrithio antitumor agents |
| US5767285A (en) | 1994-06-03 | 1998-06-16 | American Cyanamid Company | Linkers useful for the synthesis of conjugates of methyltrithio antitumor agents |
| US5543152A (en) | 1994-06-20 | 1996-08-06 | Inex Pharmaceuticals Corporation | Sphingosomes for enhanced drug delivery |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5591584A (en) | 1994-08-25 | 1997-01-07 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5597909A (en) | 1994-08-25 | 1997-01-28 | Chiron Corporation | Polynucleotide reagents containing modified deoxyribose moieties, and associated methods of synthesis and use |
| US5580731A (en) | 1994-08-25 | 1996-12-03 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| WO1996007754A1 (en) | 1994-09-02 | 1996-03-14 | The Scripps Research Institute | Methods for producing antibody libraries using universal or randomized immunoglobulin light chains |
| WO1996007321A1 (en) | 1994-09-06 | 1996-03-14 | The Uab Research Foundation | Methods for modulating protein function in cells using intracellular antibody homologues |
| US5591721A (en) | 1994-10-25 | 1997-01-07 | Hybridon, Inc. | Method of down-regulating gene expression |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5512295A (en) | 1994-11-10 | 1996-04-30 | The Board Of Trustees Of The Leland Stanford Junior University | Synthetic liposomes for enhanced uptake and delivery |
| US5663149A (en) | 1994-12-13 | 1997-09-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide heterocyclic and halophenyl amides |
| US5792747A (en) | 1995-01-24 | 1998-08-11 | The Administrators Of The Tulane Educational Fund | Highly potent agonists of growth hormone releasing hormone |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| US5641870A (en) | 1995-04-20 | 1997-06-24 | Genentech, Inc. | Low pH hydrophobic interaction chromatography for antibody purification |
| WO1996033735A1 (en) | 1995-04-27 | 1996-10-31 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO1996034096A1 (en) | 1995-04-28 | 1996-10-31 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5712374A (en) | 1995-06-07 | 1998-01-27 | American Cyanamid Company | Method for the preparation of substantiallly monomeric calicheamicin derivative/carrier conjugates |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US5652356A (en) | 1995-08-17 | 1997-07-29 | Hybridon, Inc. | Inverted chimeric and hybrid oligonucleotides |
| WO1997030087A1 (en) | 1996-02-16 | 1997-08-21 | Glaxo Group Limited | Preparation of glycosylated antibodies |
| WO1997033551A2 (en) | 1996-03-15 | 1997-09-18 | Millennium Pharmaceuticals | Compositions and methods for the diagnosis, prevention, and treatment of neoplastic cell growth and proliferation |
| US6919436B2 (en) | 1996-08-30 | 2005-07-19 | Upfront Chromatography A/S | Isolation of proteins |
| US20050176122A1 (en) | 1996-08-30 | 2005-08-11 | Upfront Chromatography A/S | Isolation of proteins |
| US6136958A (en) * | 1996-09-30 | 2000-10-24 | Genentech, Inc. | Antibodies to vertebrate smoothened proteins |
| WO1998024893A2 (en) | 1996-12-03 | 1998-06-11 | Abgenix, Inc. | TRANSGENIC MAMMALS HAVING HUMAN IG LOCI INCLUDING PLURAL VH AND Vλ REGIONS AND ANTIBODIES PRODUCED THEREFROM |
| WO1998039352A1 (en) | 1997-03-07 | 1998-09-11 | Takeshi Imanishi | Novel bicyclonucleoside and oligonucleotide analogues |
| US20050026229A1 (en) | 1997-03-10 | 2005-02-03 | The Regents Of The University Of California | PSCA antibodies and hybridomas producing them |
| US20050100546A1 (en) | 1997-05-05 | 2005-05-12 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| US20060183887A1 (en) | 1997-05-05 | 2006-08-17 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| WO1998058964A1 (en) | 1997-06-24 | 1998-12-30 | Genentech, Inc. | Methods and compositions for galactosylated glycoproteins |
| WO1999014226A2 (en) | 1997-09-12 | 1999-03-25 | Exiqon A/S | Bi- and tri-cyclic nucleoside, nucleotide and oligonucleotide analogues |
| WO1999022764A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US7189826B2 (en) | 1997-11-24 | 2007-03-13 | Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
| US7087409B2 (en) | 1997-12-05 | 2006-08-08 | The Scripps Research Institute | Humanization of murine antibody |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| WO1999051642A1 (en) | 1998-04-02 | 1999-10-14 | Genentech, Inc. | Antibody variants and fragments thereof |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| WO2000042072A2 (en) | 1999-01-15 | 2000-07-20 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US20060059575A1 (en) | 1999-03-30 | 2006-03-16 | Japan Tobacco, Inc. | Method for preparing monoclonal antibody |
| WO2000061739A1 (en) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| US6656730B1 (en) | 1999-06-15 | 2003-12-02 | Isis Pharmaceuticals, Inc. | Oligonucleotides conjugated to protein-binding drugs |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| WO2001029246A1 (en) | 1999-10-19 | 2001-04-26 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing polypeptide |
| US20020038086A1 (en) | 2000-07-27 | 2002-03-28 | Hynynen Kullervo H. | Blood-brain barrier opening |
| US20020065259A1 (en) | 2000-08-30 | 2002-05-30 | Schatzberg Alan F. | Glucocorticoid blocking agents for increasing blood-brain barrier permeability |
| US20030115614A1 (en) | 2000-10-06 | 2003-06-19 | Yutaka Kanda | Antibody composition-producing cell |
| WO2002031140A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions |
| US20020164328A1 (en) | 2000-10-06 | 2002-11-07 | Toyohide Shinkawa | Process for purifying antibody |
| US20030073713A1 (en) | 2000-10-30 | 2003-04-17 | Schoenhard Grant L. | Inhibitors of ABC drug transporters at the blood-brain barrier |
| US20030083299A1 (en) | 2000-11-04 | 2003-05-01 | Ferguson Ian A. | Non-invasive delivery of polypeptides through the blood-brain barrier |
| US20030104402A1 (en) | 2001-01-23 | 2003-06-05 | University Of Rochester | Methods of producing or identifying intrabodies in eukaryotic cells |
| WO2002088172A2 (en) | 2001-04-30 | 2002-11-07 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US20040131692A1 (en) | 2001-05-05 | 2004-07-08 | Joerg Kreuter | Nanoparticles made of protein with coupled apolipoprotein e for penetration of the blood-brain barrier and methods for the production thereof |
| US7078492B2 (en) | 2001-05-16 | 2006-07-18 | Abgenix, Inc. | Human antipneumococcal antibodies from non-human animals |
| US20030129186A1 (en) | 2001-07-25 | 2003-07-10 | Biomarin Pharmaceutical Inc. | Compositions and methods for modulating blood-brain barrier transport |
| WO2003011878A2 (en) | 2001-08-03 | 2003-02-13 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| US7153507B2 (en) | 2001-08-23 | 2006-12-26 | Genmab A/S | Human antibodies specific for interleukin 15 (IL-15) |
| US20030157108A1 (en) | 2001-10-25 | 2003-08-21 | Genentech, Inc. | Glycoprotein compositions |
| US20040093621A1 (en) | 2001-12-25 | 2004-05-13 | Kyowa Hakko Kogyo Co., Ltd | Antibody composition which specifically binds to CD20 |
| US20050124533A1 (en) | 2002-02-27 | 2005-06-09 | Schatzberg Alan F. | Glucocorticoid blocking agents for increasing blood-brain barrier permeability stan-261con |
| US20030162695A1 (en) | 2002-02-27 | 2003-08-28 | Schatzberg Alan F. | Glucocorticoid blocking agents for increasing blood-brain barrier permeability |
| WO2003077945A1 (en) | 2002-03-14 | 2003-09-25 | Medical Research Council | Intracellular antibodies |
| US20040110704A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells of which genome is modified |
| WO2003084570A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | DRUG CONTAINING ANTIBODY COMPOSITION APPROPRIATE FOR PATIENT SUFFERING FROM FcϜRIIIa POLYMORPHISM |
| US20040110282A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Cells in which activity of the protein involved in transportation of GDP-fucose is reduced or lost |
| US20040109865A1 (en) | 2002-04-09 | 2004-06-10 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition-containing medicament |
| WO2003085119A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | METHOD OF ENHANCING ACTIVITY OF ANTIBODY COMPOSITION OF BINDING TO FcϜ RECEPTOR IIIa |
| US20040132140A1 (en) | 2002-04-09 | 2004-07-08 | Kyowa Hakko Kogyo Co., Ltd. | Production process for antibody composition |
| WO2003085107A1 (en) | 2002-04-09 | 2003-10-16 | Kyowa Hakko Kogyo Co., Ltd. | Cells with modified genome |
| EP1391213A1 (en) | 2002-08-21 | 2004-02-25 | Boehringer Ingelheim International GmbH | Compositions and methods for treating cancer using maytansinoid CD44 antibody immunoconjugates and chemotherapeutic agents |
| US20050014934A1 (en) | 2002-10-15 | 2005-01-20 | Hinton Paul R. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20040204354A1 (en) | 2002-12-03 | 2004-10-14 | Thomas Nelson | Artificial low-density lipoprotein carriers for transport of substances across the blood-brain barrier |
| WO2004056312A2 (en) | 2002-12-16 | 2004-07-08 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| US20060258841A1 (en) | 2003-01-17 | 2006-11-16 | Josef Michl | Pancreatic cancer associated antigen, antibody thereto, and diagnostic and treatment methods |
| WO2004092219A2 (en) | 2003-04-10 | 2004-10-28 | Protein Design Labs, Inc | Alteration of fcrn binding affinities or serum half-lives of antibodies by mutagenesis |
| US20050287149A1 (en) | 2003-05-21 | 2005-12-29 | Medarex, Inc. | Human monoclonal antibodies against bacillus anthracis protective antigen |
| US20050089473A1 (en) | 2003-09-10 | 2005-04-28 | Cedars-Sinai Medical Center | Potassium channel mediated delivery of agents through the blood-brain barrier |
| WO2005035586A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Fused protein composition |
| WO2005035778A1 (en) | 2003-10-09 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | PROCESS FOR PRODUCING ANTIBODY COMPOSITION BY USING RNA INHIBITING THE FUNCTION OF α1,6-FUCOSYLTRANSFERASE |
| US20050123546A1 (en) | 2003-11-05 | 2005-06-09 | Glycart Biotechnology Ag | Antigen binding molecules with increased Fc receptor binding affinity and effector function |
| WO2005053742A1 (en) | 2003-12-04 | 2005-06-16 | Kyowa Hakko Kogyo Co., Ltd. | Medicine containing antibody composition |
Non-Patent Citations (310)
| Title |
|---|
| "Cancer: Principles and Practice of Oncology", 1993, J.B. LIPPINCOTT COMPANY |
| "Cell and Tissue Culture: Laboratory Procedures", 1993, J. WILEY AND SONS |
| "Cell Biology: A Laboratory Notebook", 1998, ACADEMIC PRESS |
| "Methods in Molecular Biology", vol. 178, 2001, HUMAN PRESS, pages: 1 - 37 |
| "Monoclonal Antibodies: A Practical Approach", 2000, OXFORD UNIVERSITY PRESS |
| "OLIGODEOXYNUCLEOTIDES AS ANTISENSE INHIBITORS OF GENE EXPRESSION", 1988, CRC PRESS |
| "Short Protocols in Molecular Biology", 1999, WILEY AND SONS |
| "The Antibodies", 1995, HARWOOD ACADEMIC PUBLISHERS |
| "THE CONCISE ENCYCLOPEDIA OF POLYMER SCIENCE AND ENGINEERING", 1990, JOHN WILEY & SONS, pages: 858 - 859 |
| A. L. LEHNINGER: "Biochemistry, second ed.,", 1975, WORTH PUBLISHERS, pages: 73 - 75 |
| A. MOLCKOVSKY; L.L. SIU, J. HEMATOL. ONCOL., vol. 1, 2008, pages 20 |
| ANDERSON ET AL., SCIENCE, vol. 256, 1992, pages 808 - 813 |
| BALDWIN ET AL., LANCET, 15 March 1986 (1986-03-15), pages 603 - 05 |
| BARBAS ET AL., PROC NAT. ACAD. SCI. USA, vol. 91, 1994, pages 3809 - 3813 |
| BARBAS ET AL., PROC. NATL. ACAD. SCI USA, vol. 88, 1991, pages 7978 - 7982 |
| BARBAS ET AL., PROC. NATL. ACAD. SCI. USA, vol. 88, 1991, pages 7978 - 7982 |
| BARBAS ET AL., PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 4457 - 4461 |
| BARNES ET AL., ANAL. BIOCHEM., vol. 102, 1980, pages 255 |
| BASS ET AL., PROTEINS, vol. 8, 1990, pages 309 - 314 |
| BEHRSTOCK ET AL., GENE THER., 15 December 2005 (2005-12-15) |
| BOBO ET AL., PROC. NATL. ACAD. SCI. USA, vol. 91, 1994, pages 2076 - 2080 |
| BOCRNCR ET AL., J. IMMUNOL., vol. 147, no. 1, 1991, pages 86 - 95 |
| BOERNER ET AL., J. IMMUNOL., vol. 147, 1991, pages 86 |
| BRENNAN ET AL., SCIENCE, vol. 229, 1985, pages 81 |
| BRODEUR ET AL.: "Monoclonal Antibody Production Techniques and Applications", 1987, MARCEL DEKKER, INC., pages: 51 - 63 |
| BRUGGEMANN ET AL., YEAR IN LMMUNOL., vol. 7, 1993, pages 33 |
| BRUGGEMANN, M. ET AL., J. EXP. MED., vol. 166, 1987, pages 1351 - 1361 |
| BRUGGERMANN ET AL., YEAR IN IMMUNOL., vol. 7, 1993, pages 33 |
| C. A. JANEWAY; P. TRAVERS: "Immunobiology", 1997 |
| C. M. RUDIN ET AL., N. ENGL. J. MED., vol. 361, 2009, pages 1173 - 1178 |
| CAPEL ET AL., IMMUNOMETHODS, vol. 4, 1994, pages 25 - 34 |
| CARLSSON ET AL., BIOCHEM. J., vol. 173, 1978, pages 723 - 737 |
| CARTER ET AL., BIO/TECHNOLOGY, vol. 10, 1992, pages 163 - 167 |
| CARTER ET AL., BIOLTECHNOLOGY, vol. 10, 1992, pages 163 - 167 |
| CARTER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 4285 |
| CHANG J.T. ET AL., MOL. CELL, vol. 34, 2009, pages 104 - 114 |
| CHARI ET AL., CANCER RESEARCH, vol. 52, 1992, pages 127 - 131 |
| CHARLES M RUDIN ET AL: "Treatment of Medulloblastoma with Hedgehog Pathway Inhibitor GDC-0449", NEW ENGLAND JOURNAL OF MEDICINE, MASSACHUSETTS MEDICAL SOCIETY, BOSTON, MA, US, vol. 361, 17 September 2009 (2009-09-17), pages 1173 - 1178, XP007915450, ISSN: 1533-4406, [retrieved on 20090902] * |
| CHARLES M. RUDIN ET AL., N. ENGL. J. MED., 2009 |
| CHARLES M. RUDIN ET AL., N. ENGL. J. MED., vol. 361, 2009, pages 1173 - 1178 |
| CHARLTON: "Methods in Molecular Biology", vol. 248, 2003, HUMANA PRESS, pages: 245 - 254 |
| CHATAL: "Monoclonal Antibodies in Immunoscintigraphy", 1989, CRC PRESS |
| CHEN ET AL., J. MOL. BIOL., vol. 293, 1999, pages 865 - 881 |
| CHEN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 96, 1999, pages 4325 - 4329 |
| CHEN J.K. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 99, 2002, pages 14071 - 14076 |
| CHO A. ET AL., DEV. BIOL., vol. 321, 2008, pages 27 - 39 |
| CHOTHIA ET AL., J. MOL. BIOL., vol. 196, 1987, pages 90 1 |
| CHOTHIA; LESK, J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| CLACKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLYNES ET AL., PNAS (USA), vol. 95, 1998, pages 652 - 656 |
| CLYNES ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 95, 1998, pages 652 - 656 |
| COLE ET AL., MONOCLONAL ANTIBODIES AND CANCER THERAPY, 1985, pages 77 |
| COONEY ET AL., SCIENCE, vol. 241, 1988, pages 456 |
| CRAGG, M.S. ET AL., BLOOD, vol. 101, 2003, pages 1045 - 1052 |
| CRAGG, M.S.; M.J. GLENNIE, BLOOD, vol. 103, 2004, pages 2738 - 2743 |
| CREIGHTON C.J., ONCOGENE, vol. 26, 2007, pages 4648 - 4655 |
| CUNNINGHAM; WELLS, SCIENCE, vol. 244, 1989, pages 1081 - 1085 |
| D. CATTY.,: "Antibodies: A Practical Approach", 1988, IRL PRESS |
| DAËRON, ANNU. REV. IMMUNOL., vol. 15, 1997, pages 203 - 234 |
| DE HAAS ET AL., J. LAB. CLIN. MED., vol. 126, 1995, pages 330 - 41 |
| DELLOVADE, T. ET AL., ANNU. REV. NEUROSCI., vol. 29, 2006, pages 539 |
| DERVAN ET AL., SCIENCE, vol. 251, 1991, pages 1360 |
| DIEFFENBACH ET AL.: "PCR PRIMER: A LABORATORY MANUAL", 1995, COLD SPRING HARBOR LABORATORY PRESS |
| DORONINA ET AL., NATURE BIOTECHNOL., vol. 21, no. 7, 2003, pages 778 - 784 |
| DORONINA, NAT BIOTECHNOL, vol. 21, no. 7, 2003, pages 778 - 784 |
| DRUGS OF THE FUTURE, vol. 25, no. 7, 2000, pages 686 |
| DUNCAN; WINTER, NATURE, vol. 322, 1988, pages 738 - 40 |
| E. HARLOW; D. LANE: "Using Antibodies: A Laboratory Manual", 1999, COLD SPRING HARBOR LABORATORY PRESS |
| E. SCHRODER; K. LUBKE: "The Peptides", vol. 1, 1965, ACADEMIC PRESS, pages: 76 - 136 |
| EMBLETON ET AL., NUCL. ACIDS RES., vol. 20, 1992, pages 3831 - 3837 |
| ENGELMAN, J.A; J. SETTLEMAN, CTIRR. OPIN. GENET. DEV., vol. 18, no. 1, 2008, pages 73 - 79 |
| ENGLISCH ET AL.: "ANGEWANDTE CHEMIE, INTERNATIONAL EDITION", vol. 30, 1991, WILEY-VCH, pages: 613 |
| EVEN ET AL., TRENDS IN BIOTECHNOLOGY, vol. 24, no. 3, 2006, pages 105 - 108 |
| FELLOUSE, PROC. NATL. ACAD. SCI. USA, vol. 101, no. 34, 2004, pages 12467 - 12472 |
| FISHWILD ET AL., NATURE BIOTECHNOL., vol. 14, 1996, pages 845 - 851 |
| FLEER ET AL., BIOLTECHNOLOGY, vol. 9, 1991, pages 968 - 975 |
| FRAKER ET AL., BIOCHEM. BIOPHYS. RES. COMMUN., vol. 80, 1978, pages 49 - 57 |
| FRANEK, TRENDS IN MONOCLONAL ANTIBODY RESEARCH, 2005, pages 111 - 122 |
| FRANK-KAMENETSKY M. ET AL., J. BIOL., vol. 1, 2002, pages 10 |
| GAZZANO-SANTORO ET AL., J. IMMUNOL. METHODS, vol. 202, 1996, pages 163 |
| GEOGHEGAN; STROH, BIOCONJUGATE CHEM., vol. 3, 1992, pages 138 - 146 |
| GERNGROSS, NAT. BIOTECH., vol. 22, 2004, pages 1409 - 1414 |
| GHETIE ET AL., NATURE BIOTECHNOLOGY, vol. 15, no. 7, 1997, pages 637 - 640 |
| GHETIE; WARD, IMMUNOL. TODAY, vol. 18, no. 12, 1997, pages 592 - 598 |
| GILL ET AL., NATURE MED., vol. 9, 2003, pages 589 - 595 |
| GODING: "Monoclonal Antibodies: Principles and Practice", 1986, ACADEMIC PRESS, pages: 59 - 103 |
| GOODRICH, L.V. ET AL., SCIENCE, vol. 277, no. 5329, 1997, pages 1109 - 1113 |
| GRACHTCHOUK M. ET AL., NAT GENET., vol. 24, 2000, pages 216 - 217 |
| GRAHAM ET AL., J. GEN VIROL., vol. 36, 1977, pages 59 |
| GRAM, PROC. NATL. ACAD. SCI USA, vol. 89, 1992, pages 3576 - 3580 |
| GRIFFITHS ET AL., EMBO J, vol. 12, 1993, pages 725 - 734 |
| GRUBER ET AL., J. IMMUNOL., vol. 152, 1994, pages 5368 |
| GUSS ET AL., EMBO.J., vol. 5, 1986, pages 1567 - 1575 |
| GUYER ET AL., J. IMMUNOL., vol. 117, 1976, pages 587 |
| GUYER ET AL., J. LMMUNOL., vol. 117, 1976, pages 587 |
| H. LI; J. RUAN; R. DURBIN, GENOME RES., vol. 18, 2008, pages 1851 |
| HAM ET AL., METH. ENZ., vol. 58, 1979, pages 44 |
| HAMERS-CASTERMAN ET AL., NATURE, vol. 363, 1993, pages 446 - 448 |
| HAMMERLING ET AL.: "Monoclonal Antibodies and T-Cell Hybridomas", 1981, ELSEVIER, pages: 563 - 681 |
| HAN Y.G. ET AL., NAT MED., vol. 15, 2009, pages 1062 - 1065 |
| HARLOW ET AL.: "Antibodies: A Laboratory Manual", 1988, COLD SPRING HARBOR LABORATORY PRESS |
| HARRIS, BIOCHEM. SOC. TRANSACTIONS, vol. 23, 1995, pages 1035 - 1038 |
| HARTMANN W. ET AL., CLIN. CANCER RES., vol. 12, 2005, pages 3019 - 27 |
| HAWKINS ET AL., J. MOL. BIOL., vol. 226, 1992, pages 889 - 896 |
| HELLSTROM, I ET AL., PROC. NAT'L ACAD. SCI. USA, vol. 82, 1985, pages 1499 - 1502 |
| HELLSTROM, I. ET AL., PROC. NAT'1 ACAD. SCI. USA, vol. 83, 1986, pages 7059 - 7063 |
| HINMAN ET AL., CANCCR RCSCARCH, vol. 53, 1993, pages 3336 - 3342 |
| HINMAN ET AL., CANCER RES., vol. 53, 1993, pages 3336 - 3342 |
| HINTON ET AL., J. BIOL. CHEM., vol. 279, no. 8, 2004, pages 6213 - 6216 |
| HOGREFE ET AL., GENE, vol. 128, 1993, pages 119 - 126 |
| HOLLINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HONGO ET AL., HYBRIDOMA, vol. 14, no. 3, 1995, pages 253 - 260 |
| HOOGENBOOM ET AL., NUCL. ACIDS RES., vol. 19, 1991, pages 4133 - 4137 |
| HOOGENBOOM; WINTER, J. MOL. BIOI., vol. 227, 1992, pages 381 - 388 |
| HOOGENBOOM; WINTER, J. MOL. BIOL., vol. 227, 1991, pages 381 |
| HOOGENBOOM; WINTER, J. MOL. BIOL., vol. 227, 1992, pages 381 - 388 |
| HUDSON ET AL., NAT. MED., vol. 9, 2003, pages 129 - 134 |
| HURLE; GROSS, CURR. OP. BIOTECH., vol. 5, 1994, pages 428 - 433 |
| I. FRESHNEY: "Animal Cell CultureR.", 1987 |
| IDUSOGIE ET AL., J LMMUNOL., vol. 164, 2000, pages 4178 - 4184 |
| IDUSOGIE ET AL., J. IMMUNOL., vol. 164, 2000, pages 4178 - 4184 |
| J. E. COLIGAN ET AL.,: "Current Protocols in Immunology", 1991 |
| J. M. MILLER AND M. P. CALOS,: "Gene Transfer Vectors for Mammalian Cells", 1987 |
| J. P. MATHER; P. E. ROBERTS: "Introduction to Cell and Tissue Culture", 1998, PLENUM PRESS |
| J. T. ROMCR ET AL., CANCER CELL, vol. 6, 2004, pages 229 |
| J. XIE ET AL., NATURE, vol. 391, 1998, pages 90 |
| J.M. HYMAN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 106, no. 33, 2009, pages 14132 - 14137 |
| JACKSON ET AL., J. IMMUNOL., vol. 154, no. 7, 1995, pages 3310 - 9 |
| JAKOBOVITS ET AL., NATURE, vol. 362, 1993, pages 255 |
| JAKOBOVITS ET AL., NATURE, vol. 362, 1993, pages 255 - 258 |
| JAKOBOVITS ET AL., PROC. NATL. ACAD. SCI USA, vol. 90, 1993, pages 2551 |
| JAKOBOVITS ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 2551 |
| JOHNSON; WU: "Methods in Molecular Biology", vol. 248, 2003, HUMAN PRESS, pages: 1 - 25 |
| JONES ET AL., BIOTECHNOL., vol. 9, 1991, pages 88 - 89 |
| JONES ET AL., NATURE, vol. 321, 1986, pages 522 - 525 |
| JONES, GENETICS, vol. 85, 1977, pages 12 |
| KABANOV ET AL., FEBS LETT., vol. 259, 1990, pages 327 - 330 |
| KABAT ET AL.: "Proteins of Immunological Interest", vol. 1-3, 1991, NIH PUBLICATION 91-3242 |
| KABAT ET AL.: "Sequences of Immunological Interest", 1991, NATIONAL INSTITUTES OF HEALTH |
| KABAT ET AL.: "Sequences of Proteins o/` Immunological Interest", 1991, NATIONAL INSTITUTES OF HEALTH |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest", 1991, NATIONAL INSTITUTE OF HEALTH |
| KAM ET AL., PROC. NATL. ACAD. SCI. USA, vol. 102, 2005, pages 11600 - 11605 |
| KANDA, Y. ET AL., BIOTECHNOL. BIOENG., vol. 94, no. 4, 2006, pages 680 - 688 |
| KIM ET AL., 1. LMMUNOL., vol. 24, 1994, pages 249 |
| KIM ET AL., J. IMMUNOL., vol. 24, 1994, pages 249 |
| KIM J.K.; J.A. DIEHL, J. CELL. PHYSIOL., vol. 220, 2009, pages 292 - 296 |
| KOHLER ET AL., NATURE, vol. 256, 1975, pages 495 |
| KOHLER; MILSTEIN, NATURE, vol. 256, 1975, pages 495 - 97 |
| KONTCRMANN, METHODS, vol. 34, 2004, pages 163 - 170 |
| KOOL, M. ET AL., PLOS ONE, vol. 3, no. 8, 2008, pages E3088 |
| KOSTELNY ET AL., J. IMMUNOL., vol. 148, no. 5, 1992, pages 1547 - 1553 |
| KOZBOR, J. IMMUNOL., vol. 133, 1984, pages 3001 |
| KRAUSE, D.S.; R.A. VAN ETTEN, N. ENGL. J. MED., vol. 353, no. 2, 2005, pages 172 - 187 |
| LAMBERT, J., CURR. OPINION IN PHARMACOLOGY, vol. 5, 2005, pages 543 - 549 |
| LEE ET AL., J. IMMUNOL. METHODS, vol. 284, no. 1-2, 2004, pages 119 - 132 |
| LEE ET AL., J. MOL. BIOL., vol. 340, no. 5, 2004, pages 1073 - 1093 |
| LEE ET AL., J. MOL. BIOL., vol. 340, no. 5, 2004, pages 1073 - 93 |
| LEE ET AL., NUCL. ACIDS RES., vol. 6, 1979, pages 3073 |
| LETSINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 6553 - 6556 |
| LEUNG ET AL., TECHNIQUE, vol. 1, 1989, pages 11 - 15 |
| LI ET AL., NAT. BIOTECH., vol. 24, 2006, pages 210 - 215 |
| LI ET AL., PROC. NATL. ACAD. SCI. USA, vol. 103, 2006, pages 3557 - 3562 |
| LINDMARK ET AL., J. IMMUNOL. METH., vol. 62, 1983, pages 1 - 13 |
| LIU ET AL., PROC. NATL. ACAD. SCI. USA, vol. 93, 1996, pages 8618 - 8623 |
| LODE ET AL., CANCER RES., vol. 58, 1998, pages 2928 |
| LODE ET AL., CANCER RESEARCH, vol. 58, 1998, pages 2925 - 2928 |
| LONBERG ET AL., NATURE, vol. 368, 1994, pages 856 - 859 |
| LONBERG; HUSZAR, INTERN. REV. IMMUNOL., vol. 13, 1995, pages 65 - 93 |
| M. AUSUBEL, ET AL.: "Current Protocols in Molecular Biology", 2003 |
| M. J. GAIT,: "Oligonucleotide Synthesis", 1984 |
| M. J. MACPHERSON, B. D. HAMES AND G. R. TAYLOR: "Methods in Enzymology", 1995, ACADEMIC PRESS |
| MAIRA S-M ET AL.: "PI3K Inhibitors for Canccr Trcamcnt: Whcrc Do We Stand?", BIOCHEM. SOC. TRANS., vol. 37, 2009, pages 265 - 272 |
| MANDLER ET AL., BIOCONJUGATE CHEM., vol. 13, 2002, pages 786 - 791 |
| MANDLER ET AL., BIOORGANIC & MED. CHEM. LETTERS, vol. 10, 2000, pages 1025 - 1028 |
| MANDLER ET AL., J. NAT. CANCER INST., vol. 92, no. 19, 2000, pages 1573 - 1581 |
| MANOHARAN ET AL., ANN. NY. ACAD. SCI., vol. 660, 1992, pages 306 - 309 |
| MANOHARAN ET AL., BIOORG. MED. CHEM. LETT., vol. 3, 1993, pages 2765 - 2770 |
| MANOHARAN ET AL., BIOORG. MED. CHEM. LETT., vol. 4, 1994, pages 1053 - 1060 |
| MANOHARAN ET AL., NUCLEOSIDES & NUCLEOTIDES, vol. 14, 1995, pages 969 - 973 |
| MANOHARAN ET AL., TETRAHEDRON LETT., vol. 36, 1995, pages 3651 - 3654 |
| MARASCO ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 7889 - 7893 |
| MARASCO, GENE THERAPY, vol. 4, 1997, pages 11 - 15 |
| MARKS ET AL., BIO/TECHNOLOGY, vol. 10, 1992, pages 779 - 783 |
| MARKS ET AL., BIOLTECHNOLOGY, vol. 10, 1992, pages 779 - 783 |
| MARKS ET AL., BIOTECHNOL., vol. 10, 1992, pages 779 - 783 |
| MARKS ET AL., J. MOL. BIOL., vol. 222, 1991, pages 581 |
| MARKS ET AL., J. MOL. BIOL., vol. 222, 1991, pages 581 - 597 |
| MARKS ET AL., J. MOL. BIOL., vol. 222, 1992, pages 581 - 597 |
| MARTIN ET AL., HELV. CHIM. ACTA, vol. 78, 1995, pages 486 - 504 |
| MATHER ET AL., ANNALS N.Y. ACAD. SCI., vol. 383, 1982, pages 44 - 68 |
| MATHER, BIOL. REPROD., vol. 23, 1980, pages 243 - 251 |
| MATSUDA ET AL., NATURE GENET., vol. 3, 1993, pages 88 - 94 |
| MILSTEIN; CUELLO, NATURE, vol. 305, 1983, pages 537 |
| MISHRA ET AL., BIOCHIM. BIOPHYS. ACTA, vol. 1264, 1995, pages 229 - 237 |
| MOLCKOVSKY, A.; L.L. SIU, J. HEMATOL. ONCOL., vol. 1, 2008, pages 20 |
| MORIMOTO ET AL., JOURNAL OF BIOCHEMICAL AND BIOPHYSICAL METHODS, vol. 24, 1992, pages 107 - 117 |
| MORRISON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 6851 - 6855 |
| MORRISON, NATURE, vol. 368, 1994, pages 812 - 813 |
| MULLIS ET AL.,: "PCR: The Polymerase Chain Reaction", 1994 |
| MUNSON ET AL., ANAL. BIOCHEM., vol. 107, 1980, pages 220 |
| MURONE ET AL., CURR BIOL., vol. 9, 1999, pages 76 - 84 |
| MURONE M. ET AL., CURR BIOL., vol. 9, 1999, pages 76 - 84 |
| NEUBERGER, NATURE BIOTECHNOL., vol. 14, 1996, pages 826 |
| NEUWELT, E. A.: "Implication of the Blood-Brain Barrier and its Manipulation", vol. 1, 2, 1989, PLENUM PRESS |
| NI, XIANDAI MIANYIXUE, vol. 26, no. 4, 2006, pages 265 - 268 |
| NICOLAOU ET AL., ANGEW. CHEM INTL. ED. ENGL., vol. 33, 1994, pages 183 - 186 |
| NICULESCU-DUVAZ; SPRINGER, ADV. DRUG DELIV. REV., vol. 26, 1997, pages 151 - 172 |
| NIELSEN ET AL., SCIENCE, vol. 254, 1991, pages 1497 - 1500 |
| NORTHCOTT P.A. ET AL., NAT. GENET., vol. 41, 2009, pages 465 - 472 |
| OBERHAUSER, NUCL. ACIDS RES., vol. 20, 1992, pages 533 - 538 |
| OKANO, NEUROCHEM., vol. 56, 1991, pages 560 |
| OKAZAKI ET AL., J. MOL. BIOL., vol. 336, 2004, pages 1239 - 1249 |
| OLIVER T.G. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 100, 2003, pages 7331 - 7336 |
| ORLANDI ET AL., PROC. NATL. ACAD. SCI. (USA), vol. 86, 1989, pages 3833 - 3837 |
| ORUM ET AL., NUCLEIC ACIDS RES., vol. 21, 1993, pages 4491 - 4498 |
| P. FINCH, ANTIBODIES, 1997 |
| PAPANASTASSIOU ET AL., GENE THERAPY, vol. 9, 2002, pages 398 - 406 |
| PETKOVA, S.B. ET AL., INT'L. IMMUNOL., vol. 18, no. 12, 2006, pages 1759 - 1769 |
| PETTIT ET AL., ANTI-CANCER DRUG DESIGN, vol. 13, 1998, pages 243 - 277 |
| PETTIT ET AL., ANTIMICROB. AGENTS CHEMOTHER, vol. 42, 1998, pages 2961 - 2965 |
| PETTIT ET AL., J. AM. CHEM. SOC., vol. 111, 1989, pages 5463 - 5465 |
| PETTIT ET AL., J. CHEM. SOC. PERKIN TRANS., vol. 1, no. 5, 1996, pages 859 - 863 |
| PETTIT, G.R. ET AL., SYNTHESIS, 1996, pages 719 - 725 |
| PLUCKTHUN, IMMUNOL. REVS, vol. 130, 1992, pages 151 |
| PLUCKTHUN: "The Pharmacology of Monoclonal Antibodies", vol. 113, 1994, SPRINGER-VERLAG, pages: 269 - 315 |
| POGORILER J. ET AL., DEVELOPMENT, vol. 133, 2006, pages 3929 - 3937 |
| POLKINGHOM, W.R.; N.J. TARBELL, NAT. CLIN. PRACT. ONCOL., vol. 4, no. 5, 2007, pages 295 - 304 |
| PRESTA ET AL., CANCER RES., vol. 57, 1997, pages 4593 - 4599 |
| PRESTA ET AL., J. IMMUNOL., vol. 151, 1993, pages 2623 |
| PRESTA, CURR. OP. STRUCT. BIOL., vol. 2, 1992, pages 593 - 596 |
| PROC. NATL. ACAD. SCI. USA, vol. 93, 1996, pages 8618 - 8623 |
| R. L. FRESHNEY,: "Antibodies, A Laboratory Manual, and Animal Cell Culture", 1987 |
| R.T. DORSAM; J.S. GUTKIND, NAT. REV. CANCER, vol. 7, 2007, pages 79 |
| RAVETCH; KINET, ANNU. REV. IMMUNOL, vol. 9, 1991, pages 457 - 92 |
| RAVETCH; KINET, ANNU. REV. IMMUNOL., vol. 9, 1991, pages 457 - 92 |
| REMSBERG JARRETT R ET AL: "Structural analogues of smoothened intracellular loops as potent inhibitors of hedgehog pathway and cancer cell growth", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, WASHINGTON, US LNKD- DOI:10.1021/JM0705657, vol. 50, no. 18, 1 September 2007 (2007-09-01), pages 4534 - 4538, XP002496759, ISSN: 0022-2623 * |
| REYES ET AL., NATURE, vol. 297, 1982, pages 598 - 601 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 327 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 329 |
| RIPKA ET AL., ARCH. BIOCHEM. BIOPHYS., vol. 249, 1986, pages 533 - 545 |
| ROBARGE KIRK D ET AL: "GDC-0449-a potent inhibitor of the hedgehog pathway.", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS 1 OCT 2009 LNKD- PUBMED:19716296, vol. 19, no. 19, 15 August 2009 (2009-08-15), epub ahead of print, pages 5576 - 5581, XP026586282, ISSN: 1464-3405 * |
| ROBERT L YAUCH ET AL: "Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma", SCIENCE, AMERICAN ASSOCIATION FOR THE ADVANCEMENT OF SCIENCE, WASHINGTON, DC; US LNKD- DOI:10.112/SCIENCE.1179386, vol. 326, 23 October 2009 (2009-10-23), pages 572, XP007915400, ISSN: 0036-8075, [retrieved on 20090924] * |
| ROESSLER E. ET AL., HUM. MOL. GENET., vol. 14, 2005, pages 2181 - 2188 |
| ROHATGI R. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 106, 2009, pages 3196 - 201 |
| ROMER J.T. ET AL., CANCER CELL, vol. 6, 2004, pages 229 - 240 |
| ROMER, J.T. ET AL., CANCER CELL, vol. 6, no. 3, 2004, pages 229 - 240 |
| ROSSI, CURRENT BIOLOGY, vol. 4, 1994, pages 469 - 471 |
| ROWLAND ET AL., CANCER IMMUNOL. IMMUNOTHER., vol. 21, 1986, pages 183 - 87 |
| RUBIN, L.L.; F.J. DE SAUVAGE, NAT. REV. DRUG DISCOV., vol. 5, 2006, pages 1026 |
| RUDIN, C.M. ET AL., N. ENGL. J. MED., vol. 361, 2009, pages 1173 - 1178 |
| SAAL L.H. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 104, 2007, pages 7564 - 7569 |
| SAISON-BEHMOARAS ET AL., EMBO.L, vol. 10, 1991, pages 1111 - 1118 |
| SALI, A.; T.L. BLUNDELL, J. MOL. BIOL., vol. 234, 1993, pages 779 |
| SAMBROOK ET AL.: "MOLECULAR CLONING: A LABORATORY MANUAL", 1989, COLD SPRING HARBOR LABORATORY PRESS |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Manual", 2001, COLD SPRING HARBOR LABORATORY PRESS |
| SANGHVI ET AL.: "ANTISENSE RESEARCH AND APPLICATIONS", 1993, CRC PRESS, pages: 276 - 278 |
| SASTRY ET AL., PROC. NATL. ACAD. SCI. (USA), vol. 86, 1989, pages 5728 - 5732 |
| SCHIER ET AL., GENE, vol. 169, 1995, pages 147 - 155 |
| SHALABY ET AL., J. EXP. MED., vol. 175, 1992, pages 217 - 225 |
| SHEA ET AL., NUCL. ACIDS RES., vol. 18, 1990, pages 3777 - 3783 |
| SHERIFF ET AL., NATURE STRUCT. BIOL., vol. 3, 1996, pages 733 - 736 |
| SHIELDS ET AL., J. BIOL. CHEM., vol. 9, no. 2, 2001, pages 6591 - 6604 |
| SIDHU ET AL., J. MOL. BIOL., vol. 338, no. 2, 2004, pages 299 - 310 |
| SIMS ET AL., J. IMMUNOL., vol. 151, 1993, pages 2296 |
| SKERRA ET AL., CURR. OPINION IN IMMUNOL., vol. 5, 1993, pages 256 |
| STINCHCOMB ET AL., NATURE, vol. 282, 1979, pages 39 |
| SURESH ET AL., METHODS IN ENZYMOLOGY, vol. 121, 1986, pages 210 |
| SVINARCHUK ET AL., BIOCHIMIE, vol. 75, 1993, pages 49 - 54 |
| SYRIGOS; EPENETOS, ANTICANCERRE,SEARCH, vol. 19, 1999, pages 605 - 614 |
| TAIPALE, J. ET AL., NATURE, vol. 406, 2000, pages 1005 |
| TAYLOR M.D. ET AL., NAT. GENET., vol. 31, 2002, pages 306 - 310 |
| THORPE ET AL.: "Monoclonal Antibodies '84: Biological And Clinical Applications", 1985, article "Antibody Carriers Of Cytotoxic Agents In Cancer Therapy", pages: 475 - 506 |
| TOMLINSON ET AL., J. MOL. BIOL., vol. 227, 1992, pages 776 - 798 |
| TRAUNECKER ET AL., EMBOJ., vol. 10, 1991, pages 3655 |
| TUTT ET AL., J. IMMUNOL., vol. 147, 1991, pages 60 |
| URLAUB ET AL., PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4216 |
| VAN DEN BERG, BIOLTECHNOLOGY, vol. 8, 1990, pages 135 |
| VAN DIJK; VAN DE WINKEL, CURR. OPIN. PHARMACOL., vol. 5, 2001, pages 368 - 74 |
| VASWANI; HAMILTON, ANN. ALLERGY, ASTHMA & IMMUNOL., vol. 1, 1998, pages 105 - 115 |
| VERHOEYEN ET AL., SCIENCE, vol. 239, 1988, pages 1534 - 1536 |
| VITETTA ET AL., SCIENCE, vol. 238, 1987, pages 1098 |
| VIVANCO I.; C.L. SAWYERS, NAT. REV. CANCER, vol. 2, 2002, pages 489 - 501 |
| VOLLMERS; BRANDLEIN, HISTOLOGY AND HISTOPATHOLOGY, vol. 20, no. 3, 2005, pages 927 - 937 |
| VOLLMERS; BRANDLEIN, METHODS AND FINDINGS IN EXPERIMENTAL AND CLINICAL PHARMACOLOGY, vol. 27, no. 3, 2005, pages 185 - 91 |
| WANG Y. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 106, 2009, pages 2623 - 2628 |
| WARD ET AL., NATURE, vol. 341, 1989, pages 544 - 546 |
| WATERHOUSE ET AL., NUCL. ACIDS RES., vol. 21, 1993, pages 2265 - 2266 |
| WEN X. ET AL., MOL. CELL. BIOL., vol. 30, 2010, pages 1910 - 1922 |
| WETMORE C. ET AL., CANCER RES., vol. 61, 2001, pages 513 - 516 |
| WILLIAMS; WINTER, EUR..I. IMMUNOL., vol. 23, 1993, pages 1456 - 1461 |
| WILSON C.W. ET AL., PLOS ONE, vol. 4, 2009, pages E5182 |
| WINTER ET AL., ANN. REV. IMMUNOL., vol. 12, 1994, pages 433 - 455 |
| WISEMAN ET AL., BLOOD, vol. 99, no. 12, 2002, pages 4336 - 42 |
| WISEMAN ET AL., EUR. JOUR. NUCL. MED., vol. 27, no. 7, 2000, pages 766 - 77 |
| WITZIG ET AL., J. CLIN. ONCOL., vol. 20, no. 10, 2002, pages 2453 - 63 |
| WITZIG ET AL., J. CLIN. ONCOL., vol. 20, no. 15, 2002, pages 3262 - 69 |
| WONG H. ET AL., XENOBIOTICA, vol. 39, 2009, pages 850 - 861 |
| WOYKE ET AL., ANTIMICROB. AGENTS AND CHEMOTHER., vol. 45, no. 12, 2001, pages 3580 - 3584 |
| WRIGHT ET AL., TIBTECH, vol. 15, 1997, pages 26 - 32 |
| WU ET AL., NATURE BIOTECHNOLOGY, vol. 23, no. 9, 2005, pages 1137 - 1146 |
| XIE, J. ET AL., NATURE, vol. 391, 1998, pages 90 |
| XU ET AL., IMMUNITY, vol. 13, 2000, pages 37 - 45 |
| YAMANE-OHNUKI ET AL., BIOTECH. BIOENG., vol. 87, 2004, pages 614 |
| YANIV, NATURE, vol. 297, 1982, pages 17 - 18 |
| YAUCH ROBERT L ET AL: "A paracrine requirement for hedgehog signalling in cancer", NATURE (LONDON), vol. 455, no. 7211, September 2008 (2008-09-01), pages 406, XP002520120, ISSN: 0028-0836 * |
| YAZAKI; WU: "Methods in Molecular Biology", vol. 248, 2003, HUMANA PRESS, pages: 255 - 268 |
| YELTON ET AL., J. IMMUNOL., vol. 155, 1995, pages 1994 - 2004 |
| ZHAO X. ET AL., CANCER RES., vol. 64, 2004, pages 3060 - 3071 |
| ZINDY F. ET AL., CANCER RES., vol. 67, 2007, pages 2676 - 2684 |
| ZURAWEL, R.H. ET AL., GENES CHROMOSOMES CANCER, vol. 27, no. 1, 2000, pages 44 - 51 |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8481680B2 (en) | 2010-10-05 | 2013-07-09 | Genentech, Inc. | Mutant smoothened and methods of using the same |
| US9096686B2 (en) | 2010-10-05 | 2015-08-04 | Genentech, Inc. | E518A/K mutant smoothened and methods of using the same |
| WO2015116902A1 (en) | 2014-01-31 | 2015-08-06 | Genentech, Inc. | G-protein coupled receptors in hedgehog signaling |
| WO2015120075A2 (en) | 2014-02-04 | 2015-08-13 | Genentech, Inc. | Mutant smoothened and methods of using the same |
| CN107636170A (en) * | 2015-02-04 | 2018-01-26 | 健泰科生物技术公司 | Saltant type Smoothened and its application method |
| US10330683B2 (en) | 2015-02-04 | 2019-06-25 | Genentech, Inc. | Mutant smoothened and methods of using the same |
| WO2017136558A1 (en) | 2016-02-04 | 2017-08-10 | Curis, Inc. | Mutant smoothened and methods of using the same |
| CN109071625A (en) * | 2016-02-04 | 2018-12-21 | 柯瑞斯公司 | Smooth mutant and its application method |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016073276A (en) | 2016-05-12 |
| CA2772715C (en) | 2019-03-26 |
| JP5887270B2 (en) | 2016-03-16 |
| JP2013503644A (en) | 2013-02-04 |
| JP6279527B2 (en) | 2018-02-14 |
| JP2018061516A (en) | 2018-04-19 |
| US9910050B2 (en) | 2018-03-06 |
| US20160313354A1 (en) | 2016-10-27 |
| AU2010289400A1 (en) | 2012-04-12 |
| US20120282259A1 (en) | 2012-11-08 |
| DK2473522T3 (en) | 2016-11-28 |
| EP2473522A1 (en) | 2012-07-11 |
| ES2599076T3 (en) | 2017-01-31 |
| AU2010289400B2 (en) | 2014-10-23 |
| EP2473522B1 (en) | 2016-08-17 |
| US9321823B2 (en) | 2016-04-26 |
| CA2772715A1 (en) | 2011-03-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2625197B1 (en) | Mutant smoothened and methods of using the same | |
| US9910050B2 (en) | Mutant smoothened and methods of using the same | |
| US10330683B2 (en) | Mutant smoothened and methods of using the same | |
| AU2015214264B2 (en) | Mutant Smoothened and methods of using the same | |
| AU2015252098B2 (en) | Mutant smoothened and methods of using the same | |
| US20190083646A1 (en) | Mutant smoothened and methods of using the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10751758 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2772715 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012528067 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010289400 Country of ref document: AU |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010751758 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010751758 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2010289400 Country of ref document: AU Date of ref document: 20100902 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13394069 Country of ref document: US |